




药物临床试验是从药物研发到药品上市的过程中至关重要的阶段。药品注册申请人在申请药品注册上市前,需依照国家药品监管部门的要求和法定程序完成药物临床试验,而药物临床试验过程中产生的数据是证明药品安全性和有效性的重要依据。因此,药物临床试验数据合规对于药品注册申请人取得药品注册证书具有重要意义。
药物临床试验数据合规涉及的法律法规主要包括《药物临床试验质量管理规范》(以下称“GCP”)、《药品管理法》及《药品管理法实施条例》等。本文的上篇将就临床试验的基本概念、监管机构、临床试验各方职责、常见的数据合规风险以及数据合规建议进行阐述。
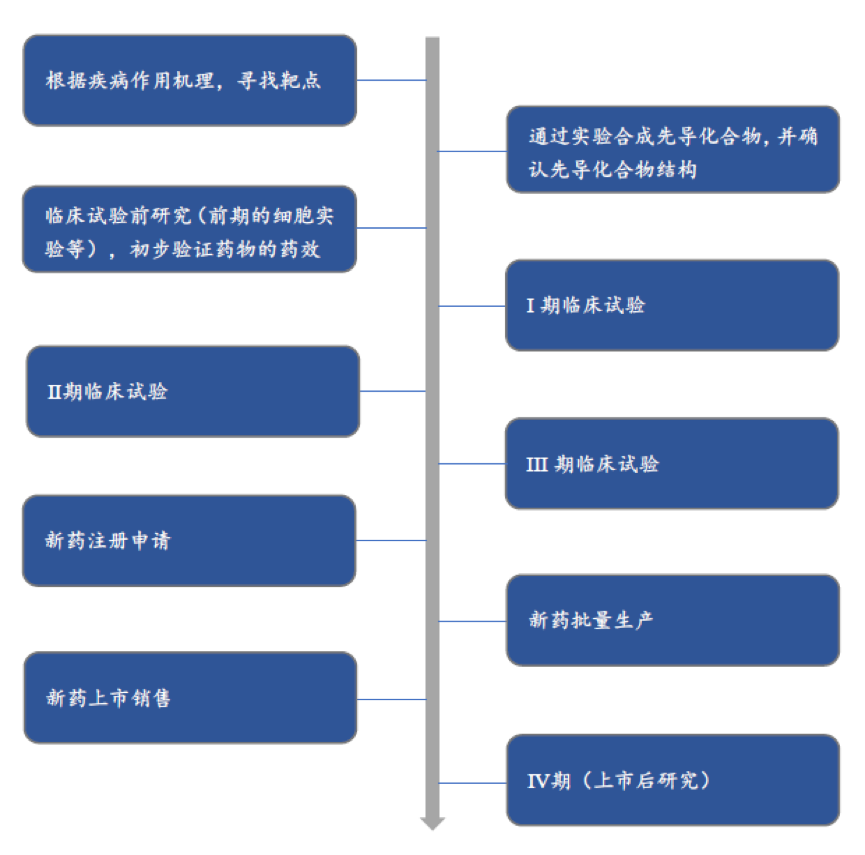
一、从药物研发到药品上市的流程
以化学药为例,其上市前一般经过如下阶段。自药物临床试验开始,医药企业(以下称“药企”)需要尤为关注其中相关的合规问题。
二、药物临床试验概述
临床试验(Clinical Trial):一般指以人体(患者或健康受试者)为对象的试验,意在发现或验证某种试验药物的临床医学、药理学以及其他药效学作用、不良反应,或者试验药物的吸收、分布、代谢和排泄,以确定药物的疗效与安全性的系统性试验。临床试验的目的是为了验证试验药物的安全性和有效性。
临床试验的类别:根据试验发起的目的,临床试验可以分为:(1)以药品上市注册为目的的临床试验(Industry-Sponsored Clinical Trial, “IST”);和(2)以扩展和优化现有疗法,提高治疗水平为主要目的的临床试验(Investigator-Initiated Clinical Trial, “IIT”);前者的申办者一般为药企;后者的发起人主要为研究者(医生/科研人员)。为免疑义,本文提及的临床试验指“IST”。
临床试验的主要监管机构:各级药品监督管理局(“药监局”)和各级卫生健康委员会(“卫健委”)。临床试验开始前,申办者应当向当地药监局提交相关的临床试验资料,并获得临床试验的许可或者完成备案;在临床试验开展过程中,申办者应当向当地药监局和卫健委报告可疑且非预期严重不良反应。
临床试验的阶段:IST药物临床试验分为I、Ⅱ、III、Ⅳ期(具体详述如下)。各阶段通常按顺序进行,也可能根据药物特点开展一个或多个研究或交叉重叠进行。

三、临床试验各方职责
从事药物研发活动,开展药物临床试验(包括备案后开展的生物等效性试验(“bioequivalence experiment”)),应当在药物临床试验机构中进行,药品上市注册申请人在开展临床试验前,应当与具有医疗机构执业许可证的临床试验机构签署相关临床试验协议。在开展临床试验前,应当通过伦理委员会的审查,并取得药监局的批准或备案。
如上所述,临床试验涉及的主体众多,我们就相关主体的责任划分梳理如下:

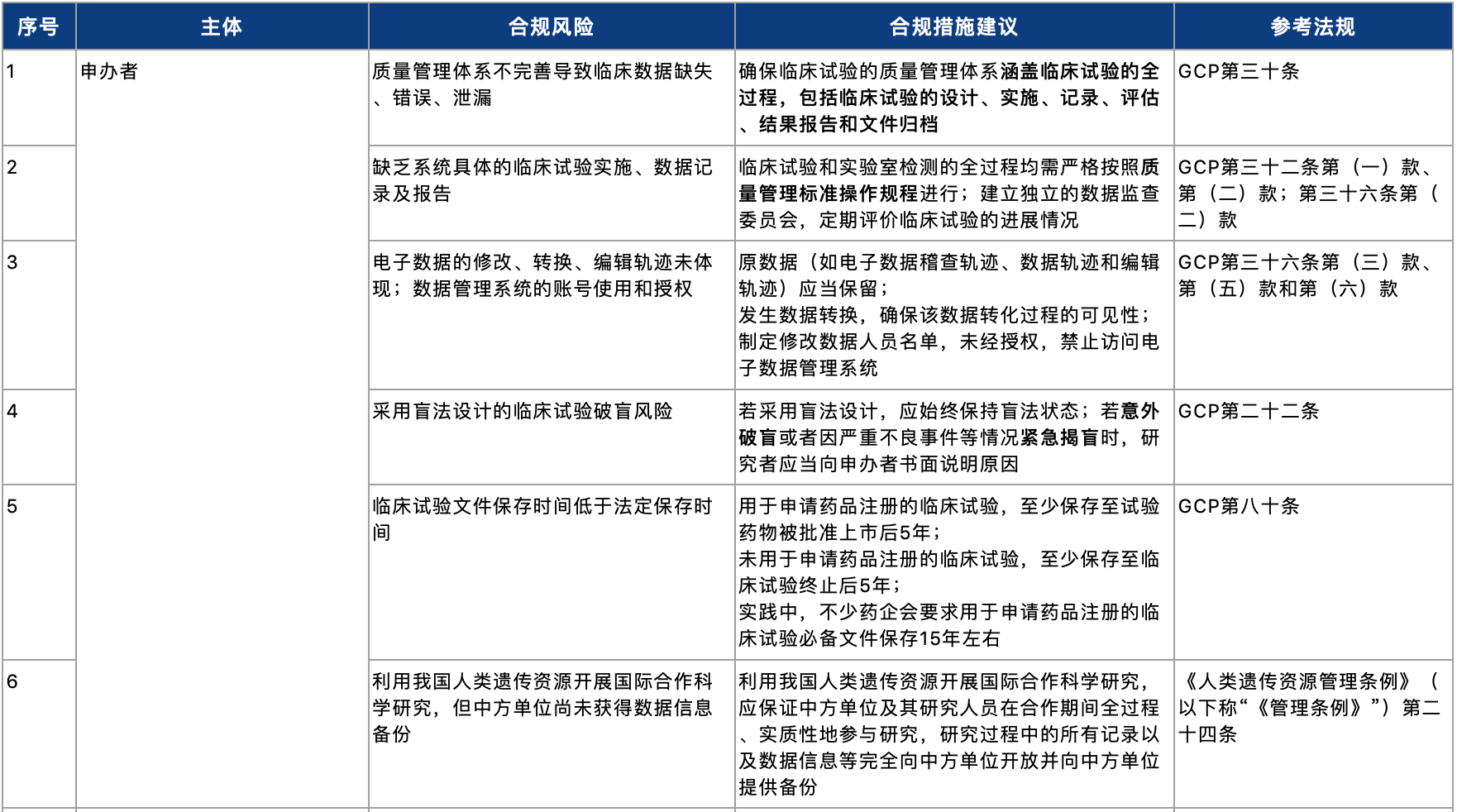
四、临床试验数据常见合规风险及合规措施
数据是临床试验极为重要的核心内容,是评估药物安全性、有效性的重要要素。完善的临床试验数据合规体系有助于药企顺利推进药物研发与药品上市,也是药物研发全生命周期的重要一环。
临床试验常见的数据不合规现象主要包括数据缺失、错误、泄漏、造假、修改、不完整、数据破盲、未经受试者同意等。
我们就临床试验不同主体在药物临床试验中常见的数据合规风险进行了如下总结,结合临床试验各主要参与主体的职责相应提出了如下的合规措施建议:

五、临床试验数据合规体系建议
考虑到药企在临床试验中可能存在前述相关数据合规风险,我们建议药企可以结合自身情况,采取如下数据合规措施,以减少后续药品上市过程中因合规风险带来的影响。
-
建立临床试验数据安全保障制度
定期对临床试验数据进行核查,建立数据核查制度与相应的质疑及修改流程。当研究者向药企书面报告严重不良事件(“SAE”)时,药企要核查、保存SAE数据,并提前制定最大限度减轻SAE影响的应对措施。
-
建立临床试验数据管理系统
为便于临床试验的药物安全性数据在药企不同部门之间以及药企与药物评审机构之间交流便利以及数据共享,应尽可能建立数据标准管理体系。为便于临床试验涉及的质量手册、质量记录等数据文件进行管理,要定时检查、维护数据管理系统的稳定性及可靠性,增强数据的可溯源性以及数据进入权限管理。
就数据管理系统设置相应的保密政策、规范,应确保数据取得合法、规范。
-
临床试验数据相关人员管理
药企应在企业内部规章制度以及与研究机构、CRO签署的协议中根据GCP的相关规定明确各方职责,并在有条件的情况下对相关人员进行定期培训(包括但不限于保密培训、标准操作规程执行培训)、评估及考核。研究机构、CRO及CRC在各方应保持对已制定标准操作规程及临床试验方案的依从性及执行力。
此外,建议在有条件的情况下,考虑对拟聘请的CRO及CRC进行尽职调查,了解其过往的任务执行情况、合规情况等。
- 临床试验数据相关合同审阅
在研发或任何外包试验的合同起草过程中,尤其注意各方主体资格、资质是否合规,里程碑事件和付款节点是否明晰,是否涵盖了数据和隐私保护内容,是否涵盖数据归属,数据收集、存储及处理等内容。
综上,建立完善的临床试验数据合规体系既保护了包括受试者在内的临床试验参与各方的权益,又有助于推进药物研发与药品上市的进程,帮助药企稳健发展。
注释:
[1] 受试者鉴认代码,指临床试验中分配给受试者以辩识其身份的唯一代码。研究者在报告受试者出现的不良事件和其他与试验有关的数据时,用该代码代替受试者姓名以保护其隐私。
[2]《个人信息保护法》第二十八条:敏感个人信息是一旦泄露或者非法使用,容易导致自然人的人格尊严受到侵害或者人身、财产安全受到危害的个人信息,包括生物识别、宗教信仰、特定身份、医疗健康、金融账户、行踪轨迹等信息,以及不满十四周岁未成年人的个人信息。


