




2025年3月19日,国家药品监督管理局(“国家药监局”)发布了《药品试验数据保护实施办法(试行)》(征求意见稿)(“《实施办法》(征)”)和《药品试验数据保护工作程序(征求意见稿)》(“《工作程序》(征)”)。药品试验数据保护是一项在多个主要医药市场已广泛开展的制度,我国此前已有相应的立法探索。本文拟就我国药品试验数据保护立法探索的沿革予以梳理,并就《实施办法》(征)及《工作程序》(征)的主要内容予以浅析,供读者参考。
一、药品数据保护制度发展历程
药品数据保护制度是一种在专利制度之外保护医药企业的制度,其使得医药企业药品在上市后对其自行取得的原始数据享有一定期限市场独占期,通过不予批准其他企业利用未披露数据进行上市申请,从而为医药企业提供额外的保护机制。药品数据保护制度起源于美国,后续又在欧盟、日本、瑞士等主要医药市场广泛实施。作为世界贸易组织(WTO)框架下的核心条约,《与贸易有关的知识产权协定》(TRIPs)即原则性地规定:各成员如要求,在涉及新型化学个体制造的药品时应保护“通过巨大努力取得的、未披露的试验数据或其他数据”并防止对其不正当的商业使用,保护这些数据不被披露[1]。我国在加入WTO后相应启动了对这一规定的立法进程。
早在2002年发布的《药品管理法实施条例》中即规定了药品试验数据保护制度,其赋予药品生产者或者销售者自获得生产、销售新型化学成份药品的许可证明文件之日起6年的数据保护期[2]。2007年10月1日生效的《药品注册管理办法》(现已修订)第20条以及《药品管理法实施条例》(2019年修订版)第34条再次重申了该项规定。
2017年5月,原国家食品药品监督管理总局发布的《关于鼓励药品医疗器械创新保护创新者权益的相关政策(征求意见稿)》提出完善药品数据保护制度,并首次提出依药品分类不同而设置不同数据保护期的规定,即就创新药、罕见病用药、儿童专用药、创新的治疗用生物制品、挑战专利成功和境外已上市但境内首仿上市的药品的数据保护期作出不同的数据保护期的规定[3]。2017年10月8日,中共中央办公厅、国务院办公厅又发布了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》。该意见明确提出要完善和落实药品试验数据保护制度,对创新药、罕见病治疗药品、儿童专用药、创新治疗用生物制品以及挑战专利成功药品注册申请人提交的自行取得且未披露的试验数据和其他数据,给予一定的数据保护期。
2018年4月25日,国家药品监督管理局办公室就曾经发布《药品试验数据保护实施办法(暂行)(征求意见稿)》(“《2018实施办法》(征)”),以更为完整的形式系统地按分类规划了药品数据保护制度,包括给予创新治疗用生物制品12年数据保护期等较大“跨度”的创新。不过,由于各种原因,《2018实施办法》(征)并未迈入实施阶段。
此后,在2022年5月9日发布的《药品管理法实施条例(修订草案征求意见稿)》中,一度将数据保护范围表述为“获批上市部分药品”。但在2024年12月修订的《药品管理法实施条例》中,其措辞仍然基本维持了2002年版的语言,即赋予药品生产者或者销售者自获得生产、销售新型化学成份药品的许可证明文件之日起6年的数据保护期。2025年初发布的《国务院办公厅关于全面深化药品医疗器械监管改革促进医药产业高质量发展的意见》也再次强调了对部分药品分类别给予一定的数据保护期。自我国建立药品数据保护制度以来,在实施细则阶段,药品数据保护仍然处于仅有原则性规定而无实施细节的状态[4]。
本次《实施办法》(征)再次基于前述立法探索,按照新的药品分类标准探索了对应的数据保护制度。
二、主要规定
(一)数据保护的对象、范围及期限
《实施办法》(征)于第3条对概念进行了界定,于第4条对受保护的数据条件进行了明确,并在第5-7条等处分别对创新药、改良型新药及仿制药品的数据保护作出了规定。
相较《2018实施办法》(征),《实施办法》(征)存在一些变化:
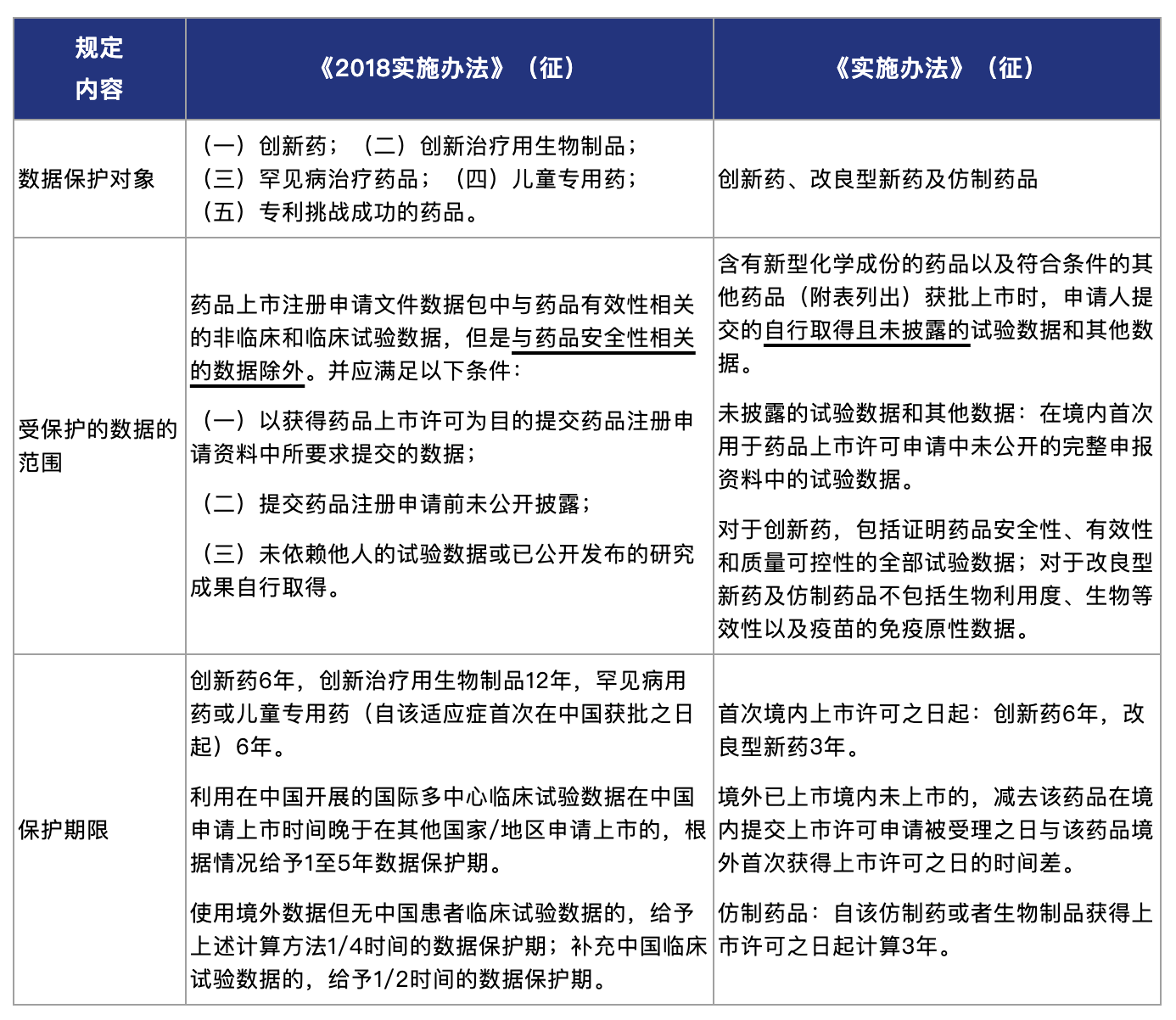
1. 数据保护的对象
从数据保护的对象上看:《实施办法》(征)在大类上将保护对象分为(1)创新药,(2)改良型新药,以及(3)首家获得批准的境外已上市境内未上市原研药品的仿制药(含境外生产药品)和生物制品。这既包含了TRIPs项下规定的“新型化学个体制造的药品”,也涵盖了近年来大量涌现的生物制品方向,并在仿制药角度有了新的突破。而首家获批的境外已上市境内未上市原研药品的仿制药(含境外生产)和生物制品在境内申请上市的过程中也可能涉及境内开展的临床试验,将其纳入数据保护范围也呼应了TRIPs中所述的对“通过巨大努力取得的”的数据的保护的要求[5]。可以说,此次类别的调整,在一定程度上也回应了目前业内对药品开发和数据保护的需求,是经过了审慎考量的决定。
不过,《实施办法》(征)不再另行设定创新治疗用生物制品的数据保护期,而将其统一放入创新药的框架内,统一给予6年的数据保护期(而非《2018实施办法》(征)中规定的12年)。《实施办法》(征)也没有再单独规定罕见病治疗药品、儿童专用药及专利挑战成功的药品的数据保护期。其实,罕见病用药、儿童专用药、生物制品等药品的数据保护已在此前原国家食品药品监督管理总局、国务院等发布的文件中提及。部分特殊类别的药品(孤儿药、儿童用药等)在美国、欧盟等主要药品市场的立法中也已体现(且期限可长于6年)。从药品的紧缺性等角度考虑,适当细化调整此类药品的数据保护期,也有助于鼓励对此类药品的研发。
2. 受保护数据的范围
从受保护数据的范围来看:以创新药为例,《2018实施办法》(征)仅纳入了与药品有效性相关的非临床和临床试验数据,排除了药品安全性数据。而《实施办法》(征)将其扩大到了证明药品安全性、有效性和质量可控性的全部试验数据。对于改良型新药和仿制药品,《实施办法》(征)的数据保护范围包括临床试验数据,但不包括生物利用度、生物等效性以及疫苗的免疫原性数据。
3. 保护期限
从受保护数据的期限来看:《实施办法》(征)总体设置了创新药6年,改良型新药3年,仿制药品3年的保护期限。在此基础上,《实施办法》(征)设置了一些额外的叠加/核减机制,其中部分机制更是此次与其他主要医药市场不同的创新机制。
(1)境外已上市境内未上市的药品保护期核减制度
《实施办法》(征)对于创新药和改良型新药设置了如下保护期的计算方式:境外已上市境内未上市的,减去该药品在境内提交上市许可申请被受理之日与该药品境外首次获得上市许可之日的时间差,数据保护期自该药品在境内获得上市许可之日起计算(即对于创新药为6年减去该时间差,对于改良型新药为3年减去该时间差)。我们理解,这一制度的初衷是希望鼓励境外新药尽早进入中国市场。我们会发现,《2018实施办法》(征)中,在核减保护期时还考虑中国临床试验数据的使用情况,而《实施办法》(征)则不再考虑这一情况。这意味着,不论中国临床试验数据的使用情况,为了尽可能在中国拥有更为长期的数据保护期,境外上市的药品就需要尽快进入中国市场。这一制度在主要医药市场中似乎还没有先例,其在实施过程中可能会成为一把双刃剑。尽管本质上讲,这一制度会推进境外上市药品尽快进入中国市场,但也可能给境外上市药品的权利人带来顾虑。毕竟,在现实的商业环境中,限于企业的全球市场规划,或者申报程序中的种种情况,反而可能导致“欲速则不达”的情况。以药品在境内提交上市许可申请被受理之日作为前述计算公式的减数,境外上市药品的权利人可能会因此而不断权衡。当然,对于境内仿制药企业而言,这一制度也可能是激励其“在起跑线前做好准备”的动力。此外,对于境外药企而言,在考虑数据保护时也需要结合其专利保护期限的情况。如果其剩余的专利保护期已少于其可获得的数据保护期,则数据保护制度对其而言仍有一定作用。
(2)首仿药(包括生物制品)保护期
《实施办法》(征)的另一大突破是,对首家获得批准的境外已上市境内未上市原研药品的仿制药(含境外生产药品)和生物制品(“首仿药”)给予3年数据保护期。并且这一制度涵盖境外生产进口和境内生产的两种渠道。同前述“在起跑线前做好准备”的理念,当境外药品未在境内上市时,境内的医药企业可以据此加速抢占先机,(某些情况下甚至是在境外权利人尚不“积极”之际)尽快为境内患者带来福利。而对境外权利人而言,为了抢占中国境内先机,其就需要尽快“出手”,阻击境内仿制药享受这一待遇。
此外,对于其他非首仿药的仿制药而言,其虽然受限于首仿药或原研药的数据保护,但其可以基于“经持有人同意”的渠道,通过许可协议等形式依赖其产生的数据开展研究。而享受数据保护的一方则可根据情况选择是否开展该等许可(如愿意共享市场),并因此获得相应的对价。
(3)新增适应症保护期
《实施办法》(征)规定,同一个批准文号的创新药,每个适应症按照注册类别分别给予数据保护。也就是说,如原有创新药的首项适应症获批后,其获得6年的数据保护期;其后如其新增一项适应症,则相应可能按改良型新药新增3年的数据保护期。其目的即在于鼓励医药企业继续开展研发,激励其拓展新的适应症的研究和改良。对于对应的仿制药对手而言,这意味着,即使该仿制药对手取得了这款创新药首项适应症的仿制药的生产许可,其将继续受限于该创新药的新适应症的数据保护。不过,在实际操作中,医师超适应症开具处方的情况是不受此限制的(医药企业也不得引导)。
(4)与部分主要医药市场的比较
与美国、欧盟等主要医药市场的立法对比,《实施办法》(征)下数据保护期的“颗粒度”仍有所不同。比如,在美国,基于Hatch-Waxman法案及相关后续法案的规定,对于新化学实体(New Chemical Exclusivity (NCE) )的保护期限为5年;如新增临床适应症、剂型等补充申请,则为3年。而对于儿科用药,设立了额外6个月的保护期限。对于孤儿药,其保护期为7年。对于“Qualified Infectious Disease Product(QIDP)”的抗生素药品,其可叠加5年的保护期[6]。此外,美国还设置了专利挑战制度下的药品数据保护的特殊规定[7]。
而在欧盟,根据Directive 2004/27/EC和Regulation (EC) No 726/2004[8],其统一设立了药品8年的数据独占期,并规定此后的2年为市场独占期(即可受理依赖该等数据的仿制药申请,但在此期间不予批准;与数据独占期合计10年)。在此基础上,原研药如在8年内获批新的临床适应症且具有显著的临床收益(Significant clinical benefit)时,前述期间可延长至共计11年。此外,孤儿药在欧盟直接享有10年的市场独占期[9],如具备符合特定的儿科适应症等情况则延长至12年[10]。故此,我们理解,此次《实施办法》(征)从统一管理的角度未就不同类型的药品数据做进一步的展开分类规定;不过,从长远的精细管理角度出发,监管部门仍可以适当考虑借鉴其他主要医药市场的立法经验。
(二)申请流程
1. 受理与审批
此次《实施办法》(征)明确了药品获得数据保护后对其他申请人申请的排除机制,即:在数据保护期届满不满1年前国家药品监督管理局药品技术审评中心(“药审中心”)不受理其他申请人依赖该受保护数据的药品上市相关申请;在届满前1年内可以受理,药审中心完成技术审评后中止审评计时,数据保护期届满后批准相关药品上市。毋庸置疑,这一制度的直接作用是减少同质化的药品研发和社会资源浪费。不过,在这一行文下,如第一家企业在届满前1年的期间内提交上市申请,但在其没有获得批准前,基于现有字面理解,其尚未被授予数据保护。在这样的“过渡期”中其他企业是否也可以提出同类申请呢?这一问题有待进一步明确。
2. 申请及公示
此次《实施办法》(征)明确:申请人拟申请数据保护的,应当在提交药品上市许可申请时同时提出数据保护申请。也就是说,申请人需要在上市申请时一并做好相应准备,“过时不候”。而申报前或者审评过程中,对数据保护相关问题存在疑问的,可与药审中心沟通交流。对于受数据保护影响暂时无法提交注册申请的药品,申请人仍可提前就相关技术问题与药审中心进行沟通交流[11]。比如,有观点指出,对相关数据是否系“依赖了其他申请人受保护数据”的情况,申请人即可据此机制与药审中心进行及时的沟通,避免事后的“一票否决”。
而对符合数据保护条件的药品,国家药监局在药品批准证明文件中标注该药品的数据保护信息。药审中心在其网站建立数据保护专栏,公布药品数据保护的相关信息。我国药品数据保护制度此前未能完全落地的一大原因即是尚未建立成熟的药品数据公示和查询平台。在美国,于FDA批准上市的药品往往会在其“橙皮书”上予以公示,公众可以查询到对应的药品信息及其数据保护的情况;而在欧盟,其也有类似的欧洲药品数据库提供相关信息。此次建立公开的公示制度,有利于医药企业及时准确了解相关数据保护情况,并据此准确合规地开展研发工作。
三、结语
此次公布的《实施办法》(征)和《工作程序》(征)等文件,吸收了我国近年来对药品数据保护制度立法不断探索的经验,借鉴了主流国际医药市场的制度经验,也在此基础上寻求创新与各方利益的平衡。在我国医药制度不断改革的时代背景下,我们期望此次相关征求意见稿发布后,主管部门可以更好地吸收各界的意见与建议,综合考虑各方的因素与权益,以求更全面地完善我国药品数据保护制度。
注释:
[1] 《与贸易有关的知识产权协定》(TRIPs)第39条第(3)款:各成员如要求,作为批准销售使用新型化学个体制造的药品或农业化学物质产品的条件,需提交通过巨大努力取得的、未披露的试验数据或其他数据,则应保护该数据,以防止不正当的商业使用。此外,各成员应保护这些数据不被披露,除非属为保护公众所必需,或除非采取措施以保证该数据不被用在不正当的商业使用中。
[2] 《药品管理法实施条例》(2002年9月15日生效版本)第35条
[3] 申请人在提交药品上市申请时,可同时提交试验数据保护申请。对批准上市的创新药,给予6年数据保护期;既属于创新药又属于罕见病用药、儿童专用药,给予10年数据保护期;属于改良型新药的罕见病用药、儿童专用药,给予3年数据保护期;属于创新的治疗用生物制品,给予10年数据保护期。挑战专利成功和境外已上市但境内首仿上市的药品给予1.5年数据保护期。欧洲药品管理局、美国和日本获准上市后1年内在中国提出上市申请和数据保护的新药,给予相应类别数据保护期;超过1年到中国提出上市申请的,按超出时间扣减数据保护期时间;扣除后不足1.5年的,给予1.5年数据保护期。数据保护期自药品批准上市之日算起。在数据保护期内,审评机构不再批准其他申请人同品种上市申请,申请人自行取得的数据除外。
[4] 在《中国-瑞士自由贸易协定》等国际贸易相关文件及磋商中,我国亦作出了不同程度的对于药品数据保护制度的承诺。
[5] 《专家视角 | 杨悦:以药品试验数据保护制度为药物研发创新注入新动能》,《中国医药报》2025年3月19日
[6] Qualified Infectious Disease Product Designation — Questions and Answers Guidance for Industry, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER)
[7] 在上市4年后,如果仿制药企业进行Paragraph IV挑战,FDA可接受该申请,但仍不能提前批准,必须等到合法程序完成(如专利诉讼)。
[8] Article 10, Directive 2004/27/EC及Article 14(11), Regulation (EC) No 726/2004
[9] Article 8, Regulation (EC) No 141/2000
[10]https://www.europarl.europa.eu/RegData/etudes/BRIE/2023/747440/EPRS_BRI(2023)747440_EN.pdf#:~:text=If%20all%20criteria%20are%20complied%20with%2C%20the%20product,a%20paediatric%20research%20and%20development%20programme%20is%20completed%29
[11] 《工作程序》(征)第13条

