




为鼓励研究和创制新药,满足临床用药需求,及时总结转化抗疫应急审评工作经验,加快创新药品的审评速度[1],国家药品监督管理局药品审评中心(以下称“CDE”)经过组织相关起草工作,于2022年2月22日发布了《药审中心加快创新药上市申请审评工作程序(试行)》(征求意见稿)(以下称“《征求意见稿》”)。《征求意见稿》明确将适用范围确定为纳入突破性治疗药物程序的创新药。本文尝试从突破性治疗药物上市申请程序相关规定切入,对本次《征求意见稿》及突破性治疗药物上市申请程序予以浅析。
一、突破性治疗药物程序的历史脉络
2020年3月30日,国家市场监督管理总局发布了新修订的《药品注册管理办法》(以下称“《药品注册管理办法》”),其于2020年7月1日起施行。《药品注册管理办法》新增了第四章“药品加快上市注册程序”,分别规定了突破性治疗药物程序、附条件批准程序、优先审评审批程序和特别审批程序。此后,国家药品监督管理局在2020年7月8日发布了《国家药监局关于发布〈突破性治疗药物审评工作程序(试行)〉等三个文件的公告》(以下称“《工作程序公告》”),对突破性治疗药物审评工作、药品附条件批准上市申请审评审批工作及药品上市许可优先审评审批工作的程序做了具体的规定。制定包括突破性治疗药物程序在内的药品加快上市注册程序,其目的是进一步完善我国药品注册审评机制,从而鼓励以临床价值为导向的药物创新。此外,药品加快上市注册程序的完善也有助于加快具有突出临床价值的临床急需药品上市并缩短新技术临床应用时间,可以弥补严重危及生命的疾病治疗手段不足的缺憾,进而更好地保障人民生命健康安全。
从国际上的经验来看,我国药品加快上市注册制度在一定程度上也借鉴了美国食品药品监督管理局(“FDA”)等国外监管机构的相关制度经验。FDA已建立包括快速通道(fast track)、突破性疗法(breakthrough therapy)、优先审评(priority review)、加速审批(accelerated approval)在内的药品加快上市制度。其中,突破性疗法通道于2012年7月设立,其更强调某种药物相比现有疗法的突破性,即突破性疗法审评旨在加快开发和审查治疗严重疾病的药物的过程,且初步临床证据表明,相关药物可能在临床显著的终点指标上显示出比现有疗法有实质性的改进。[2]在FDA的审批实践中,经认定为突破性疗法的新药,在这一制度下可以享受包括快速通道认定、FDA相关官员的密切指导以及高级审评人员参与审评工作等特殊待遇。这一通道在实践中也是一种使药物开发获得FDA青睐的“快车道”。
二、突破性治疗药物程序的适用情况
根据《药品注册管理办法》[3]及《工作程序公告》内的《突破性治疗药物审评工作程序(试行)》的规定,适用突破性治疗药物程序的情况为:药物临床试验期间,用于防治严重危及生命或者严重影响生存质量的疾病,且尚无有效防治手段或者与现有治疗手段相比有足够证据表明具有明显临床优势的创新药或者改良型新药等。同时,《突破性治疗药物审评工作程序(试行)》确认,申请适用突破性治疗药物程序的时间节点为:在Ⅰ、Ⅱ期临床试验阶段,通常不晚于Ⅲ期临床试验开展前。从现有法规及文件的表述来看,适用突破性治疗药物程序的药物需要在早期临床研发阶段就表现出相对于目前疗法的突破性特征。具体而言,《突破性治疗药物审评工作程序(试行)》对于适用突破性治疗药物程序应满足的相关条件如下:
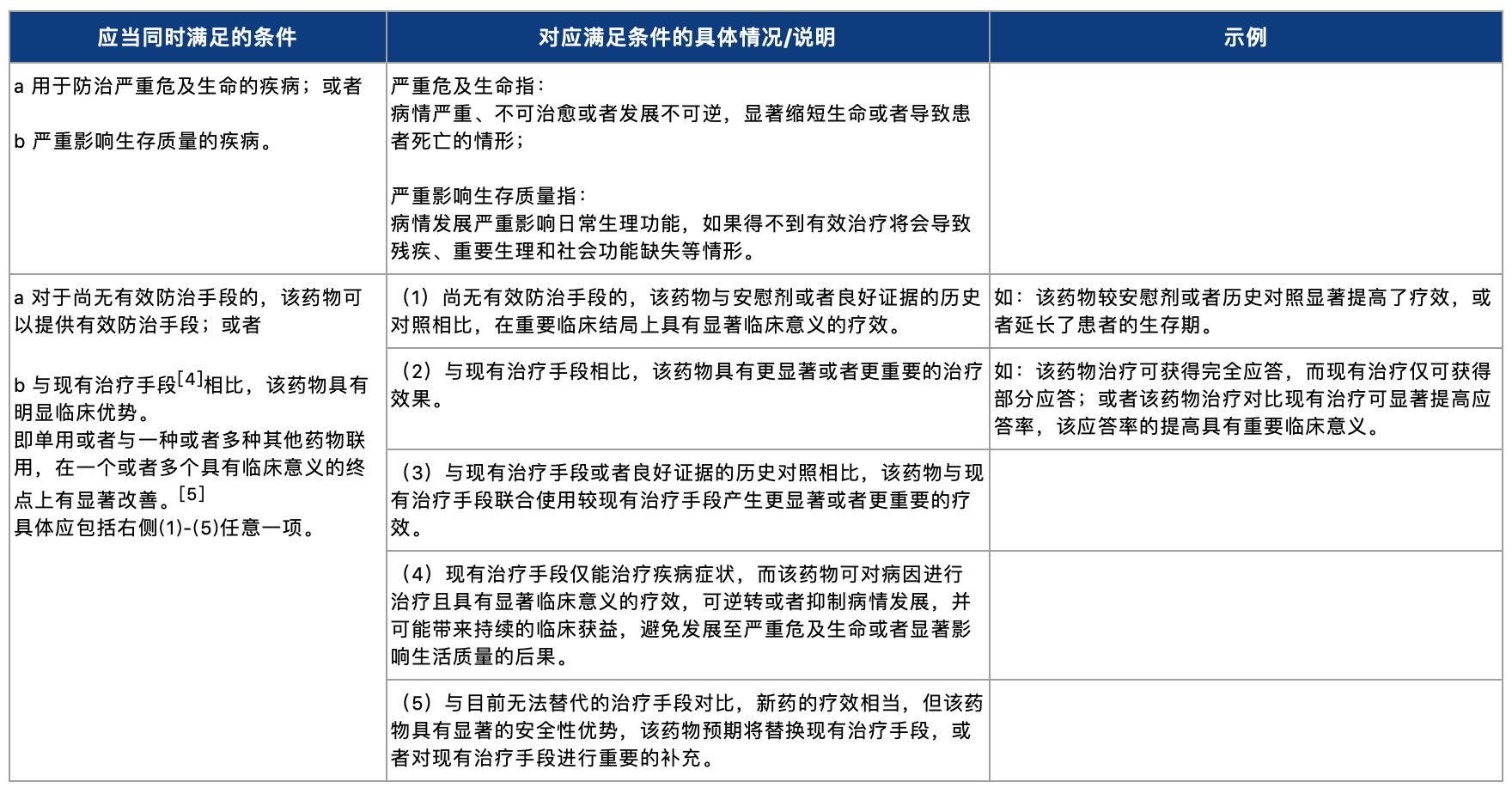
此外,值得关注的是,对于现有治疗手段的定义,《突破性治疗药物审评工作程序(试行)》明确排除了临床获益未经证实前的附条件批准上市的药品。对于适用突破性治疗药物程序的药品而言,这意味着在论证其临床优势的过程中,无需把部分尚处于附条件批准上市阶段的药品纳入需要比较的范畴。
从医药企业的实践来看,在早期临床研发阶段如能据此表现出相对于目前疗法的突破性特征,就有机会根据相关规定申请CDE将其纳入突破性药物治疗程序。而一旦纳入突破性治疗药物程序,CDE将据此优先配置相关资源,包括医药企业可以与CDE审评人员开展沟通交流、CDE基于医药企业提供的阶段性研究资料对医药企业的研究方案加强相关指导等。对于医药企业而言,这意味着其获得了CDE的“青睐”,也即获取了CDE提供的包括沟通交流及资深专家对试验指导等在内的“辅导”机会。
在现有体系下,不同程序之间可能存在一定的交叉。例如,根据《药品注册管理办法》[6]及《药品上市许可优先审评审批工作程序(试行)》,纳入突破性治疗药物程序的药品也可以申请适用优先审评审批程序。在这一制度安排下,医药企业有机会通过多种加快上市注册程序的配套衔接进一步高效、便捷地完成其上市注册流程。我们推测,在现有注册程序体系下,CDE可能倾向于通过突破性治疗药物程序更为直接、便捷地推动创新药研发及上市。本次《征求意见稿》也合理参照了其他药品加快上市程序的规定,例如,《征求意见稿》规定突破性治疗药物品种审评时限与优先审评品种时限相同(130日)。
三、突破性治疗药物程序的相关流程
(一)纳入流程
《突破性治疗药物审评工作程序(试行)》对于申请纳入突破性治疗药物程序的相关流程及时限作出了如下图所示的规定。[7]
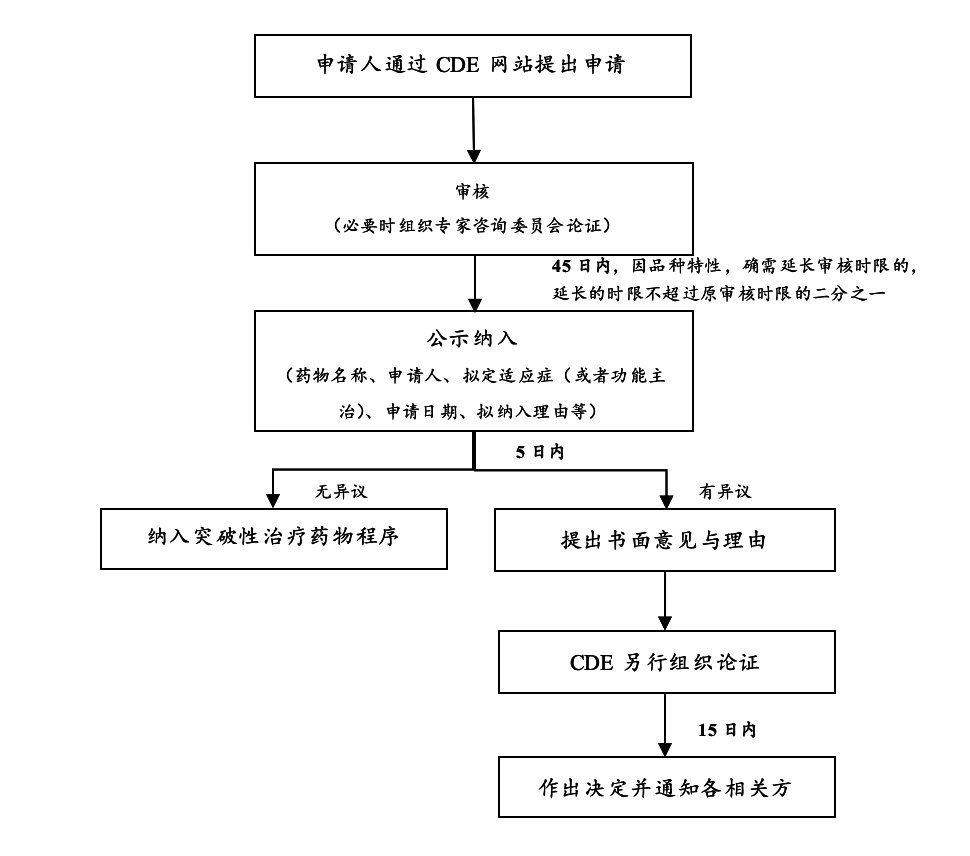
在上图所述的申请、审核、公示及纳入的程序完成后,相关药品即纳入了突破性治疗药物程序。此外,在纳入突破性治疗药物程序前,申请人也可以提出撤回申请,但需要书面说明理由。
这一流程与《征求意见稿》的规定也“无缝衔接”。《征求意见稿》规定的“加快创新药上市申请审评工作程序”无需申请人申请,纳入突破性治疗药物程序的创新药品种可自动开展该程序的后续沟通交流及审评审批工作。具体而言,该项工作程序适用于申请人在探索性临床试验完成后,已具备开展关键性临床试验条件至批准上市前。
(二)沟通交流流程
在纳入突破性治疗药物程序后,即可进入医药企业最为看重的临床试验研制指导阶段。《突破性治疗药物审评工作程序(试行)》指出,CDE应对纳入突破性治疗药物程序的药物优先配置资源进行沟通交流,这其中包括首次沟通交流、因重大安全性问题/重大技术问题而召开的会议、药物临床试验关键阶段会议以及一般性技术问题咨询等。
在沟通会议的具体安排上,《突破性治疗药物审评工作程序(试行)》与《征求意见稿》的重点有所不同:
《突破性治疗药物审评工作程序(试行)》强调了以下两项沟通:
- 首次沟通交流:在纳入突破性治疗药物程序后6个月内,申请人可以提出首次沟通交流申请,提交拟讨论的问题及相关支持性材料。未在6个月内申请首次沟通交流的,还可以在后续药物临床试验关键阶段会议申请中提交相关问题。
- 药物临床试验关键阶段会议:申请人可以在药物临床试验的关键阶段(Ⅱ期临床试验结束/Ⅲ期临床试验启动前等)向CDE提出会议申请,可以提交阶段性研究资料,CDE安排相关审评人员进行沟通交流,同时对下一步研究方案提出意见或者建议。
而《征求意见稿》在沟通交流板块中强调的是关键性临床试验相关沟通交流及上市许可申请前(pre-NDA)沟通交流。
具体而言,《征求意见稿》将关键性临床试验相关沟通的时间节点的表述做了调整,即申请人在完成前期探索性临床试验后,在关键性临床试验前和期间均可提出沟通交流申请。在这一阶段,CDE的“亲自辅导”集中体现在:
- 其与申请人会就后续沟通交流计划、阶段性研究资料提交计划、药品上市许可申请递交计划等内容进行讨论并达成一致意见;
- CDE亦将在此阶段结合具体情况,组建审评团队;
- 同时,CDE受理人员可提前介入,指导申请人按照要求整理申报资料。
而在pre-NDA沟通交流阶段,CDE对于申请人的各项“辅导”包括:
- CDE可就目前申报资料存在的问题反馈申请人,申请人可以就CDE反馈的申报材料问题再次提出沟通交流申请,滚动补充资料;
- 审评团队还需初步审核相关批件附件并与申请人沟通相关重大缺陷;
- 申请人可就提交上市许可申请后进一步提交资料的计划与CDE达成一致。
可以看出,在《征求意见稿》的安排下,申请人将进一步享受更为明确的各阶段的指导,并通过达成一致、审评团队预先审核的方式提前扫清上市申报工作中可能的障碍。此外,《征求意见稿》也明确,沟通交流的时限为30日,这也明确要求CDE对于申请人的沟通交流申请要按时答复,以尽快推进其上市审评相关流程。
(三)各项任务分配及审评安排
《征求意见稿》在核查检验、注册检验、注册核查等程序上也对申请人给予了相关便利安排,如鼓励申请人在正式申报上市前即向药品检验机构提出注册检验,如未提出注册检验的,在受理时即开具检验通知书,并在受理后10日内完成启动注册核查任务电子推送。在受理及任务分配上,其也要求CDE受理及项目管理人提前介入,审评团队负责申报前沟通交流至受理后技术审评全过程,一般包含审评人员、合规审查人员、受理人员及项目管理人员,以保证品种全链条跟进和管理。
《征求意见稿》对专业审评及综合审评提出了具体要求,并明确经过专业部门专业技术委员会讨论需要召开专家咨询会的,原则上需要10日内组织召开。
此外,《征求意见稿》在审核签发、制件送局上也提出了部分流程性要求。总而言之,《征求意见稿》主要采纳了早期介入、研审联动、滚动提交、检查检验靠前的工作理念,以达到鼓励研究和创制新药,满足临床用药需求,及时总结转化抗疫应急审评工作经验,加快创新药品的审评速度的效果。[8]
(四)终止程序
此外,根据《突破性治疗药物审评工作程序(试行)》的规定,对于纳入突破性治疗药物程序的药物临床试验,当其不再符合纳入条件时:
- 若由申请人发现,应当及时向CDE提出终止程序。
- 若由CDE发现,应当告知申请人,申请人可以在10日内提交书面说明,由CDE组织论证并在30日内作出决定。如申请人未在10日提交书面说明的,或者经论证作出决定不符合纳入条件的,CDE应当及时终止该品种的突破性治疗药物程序。
四、结语
根据CDE“纳入突破性治疗品种名单”公示,截至2022年4月13日,已有102项药品品种被纳入突破性治疗药物名单。[9]以突破性治疗药物程序为基础,为创新药完善相关加快审评工作程序,将有效鼓励医药企业在新药研发中更加注重以临床价值为导向的新药研发。我们相信,随着创新药加快上市注册程序制度的日益完善,我国的创新药研发前景将越来越好,创新药上市进程将越来越快,而医药企业也应抓紧这一机遇,充分享受制度改革带来的红利。
注释:
[1] 《药审中心加快创新药上市申请审评工作程序(试行)》(征求意见稿)起草说明
[2] “Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s).” 原文参见https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/breakthrough-therapy
[3] 《药品注册管理办法》第五十九条
[4] 《突破性治疗药物审评工作程序(试行)》指出,现有治疗手段是指在境内已批准用于治疗相同疾病的药品,或者标准治疗方法(药械组合治疗等)。通常,这些治疗手段应为当前标准治疗。
[5] 《突破性治疗药物审评工作程序(试行)》指出,具有临床意义的终点通常指与疾病发生、发展、死亡和功能等相关的终点,也可以包括经过验证的替代终点、可能预测临床获益的替代终点或者中间临床终点、安全性终点等。申请人在提出突破性治疗药物程序申请时,应当提供拟采用终点的支持性证据。
[6] 《药品注册管理办法》第六十八条
[7] 参照《〈药品注册管理办法〉学与践 | 带您快速了解药品加快上市注册程序》,作者:佟路、王海辉,载于http://www.cnpharm.com/c/2020-07-27/744858.shtml
[8] 《药审中心加快创新药上市申请审评工作程序(试行)》(征求意见稿)起草说明
[9] 原名单参见https://www.cde.org.cn/main/xxgk/listpage/da6efd086c099b7fc949121166f0130c。此外,另有1项药品目前正处于“拟突破性治疗品种”公示阶段。

