




On January 6, 2025, the U.S. Food and Drug Administration (FDA) released a draft guidance titled Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products[1] (hereinafter referred to as the “Guidance”) and sought public comment, with a 90-day comment period. We expect that the Guidance will be finalized and officially issued within this year.
Domestic and foreign enterprises that have adopted or plan to adopt artificial intelligence (AI) in the development and oversight of drugs and biological products should closely monitor the progress of this Guidance. For now, we suggest that relevant companies should promptly strategize to enhance data and AI governance frameworks, establish internal risk management mechanism, and refine work standards and operational models for regulatory submissions in alignment with the new requirements. Additionally, for activities involving FDA-regulated submissions, companies must elevate their management awareness and professional expertise in the use of AI tools, maintain active communication with regulatory authorities, and effectively manage the compliance costs associated with the proposed rules.
I. The Scope and Main Content of the Guidance
The Guidance provides recommendations to sponsors and other interested parties on the use of AI to produce information or data intended to support regulatory decision-making regarding safety, effectiveness, or quality for drugs.
Notably, the Guidance does not address the use of AI models (1) in drug discovery or (2) in applications for operational efficiencies (e.g., internal workflows, resource allocation, drafting/writing a regulatory submission) that do not impact patient safety, drug quality, or the reliability of results from a nonclinical or clinical study.
The central pillar of the Guidance is a risk-based credibility assessment framework, which is intended to help sponsors and other interested parties plan, gather, organize, and document information to establish the credibility of AI model outputs when the model is used to produce information or data intended to support regulatory decision-making.
II. Nature of the Guidance
The Guidance is non-binding in nature. FDA aims to encourage sponsors and other interested parties to adapt the Guidance to establish AI governance standards and processes tailored to their specific operational contexts and risk profiles.
While the Guidance serves as a reference resource, the FDA strongly encourages sponsors and other interested parties to initiate early engagement with FDA to align on the scope, content, and implementation measures of AI governance frameworks.
In addition, the Guidance further elaborates different options by which sponsors and other interested parties may engage with the Agency on issues related to AI model use, depending on the context of use (COU) and the specific development program. The options include requesting formal meetings and communicating with FDA’s affiliated agencies or research program coordination offices/departments like Center for Clinical Trial Innovation (C3TI), Complex Innovative Trial Design Meeting Program (CID), Drug Development Tools (DDTs), Innovative Science and Technology Approaches for New Drugs (ISTAND), Digital Health Technologies Program (DHTs), Emerging Drug Safety Technology Program (EDSTP), CDER’s Emerging Technology Program (ETP), CBER’s Technologies Team (CATT), Model-Informed Drug Development Paired Meeting Program (MIDD) and Real-World Evidence Program (RWE).
Although the FDA did not explicitly regulate or limit the specific timelines for sponsor engagement, sponsors are strongly encouraged to early engage with FDA to discuss the credibility assessment framework (detailed in Figure 1), where the FDA intends to provide interactive feedback concerning the assessment of the AI model risk (step 3) as well as the adequacy of the credibility assessment plan (step 4) based on the model risk and the COU.
III. A Risk-based Credibility Assessment Framework
Guidance is a risk-based credibility assessment framework that may be used for establishing and evaluating the credibility of an AI model for a particular COU.
Generally, this framework requires sponsors to first define the question of interest, then determine whether the AI model’s outputs will serve as the primary decision-making factor or be integrated with other evidence. After assessing the model risk, sponsors must develop a detailed plan encompassing data governance, training methodologies, performance benchmarks, and technical strategies to mitigate potential deviations. The results of the credibility assessment plan should be included in a credibility assessment report, describing any deviation from the credibility assessment plan. Ultimately, sponsors must decide whether model credibility is sufficiently established for the model risk. Improvements or enhancements to the model’s credibility level may be required before proceeding with regulatory submissions or implementation if model credibility is insufficient.
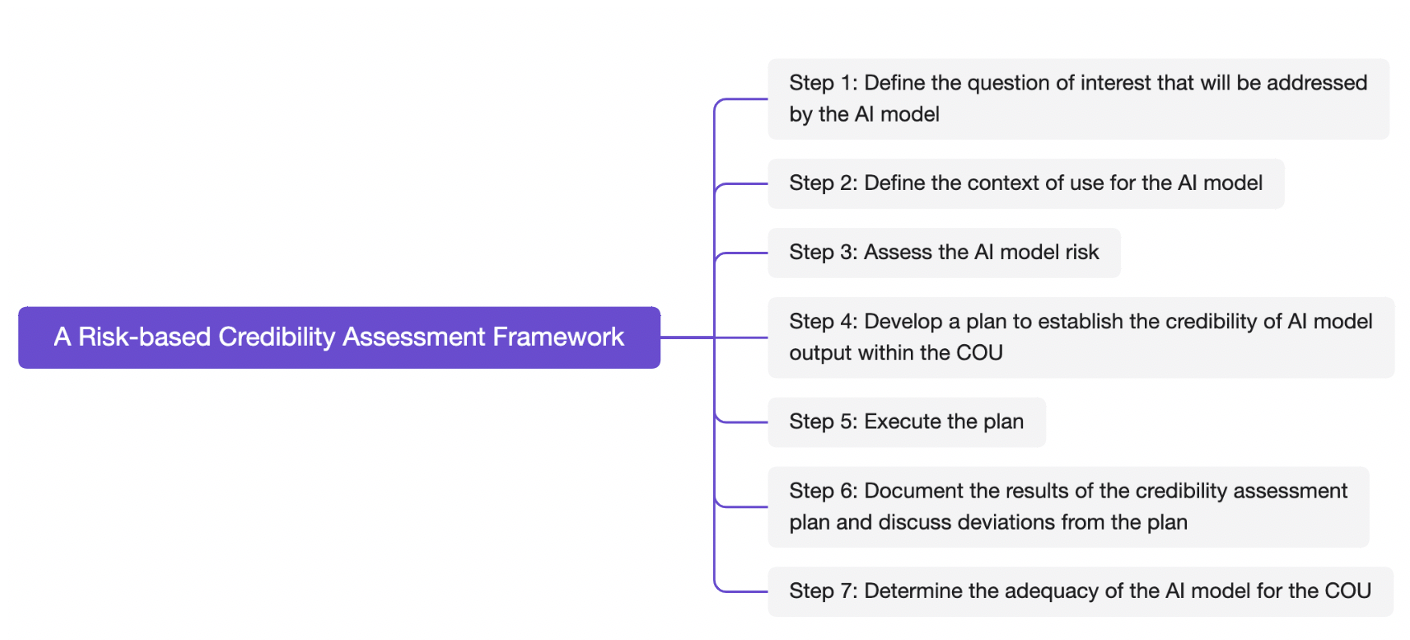
Figure 1 A Risk-based Credibility Assessment Framework
A. Step 1: Define the Question of Interest
The question of interest should describe the specific question, decision, or concern being addressed by the AI model. The Guidance provides two examples.
-
Example 1 (clinical development): Drug A is associated with a life-threatening drug-related adverse reaction. In previous trials, all participants went through 24-hour inpatient monitoring after dosing, but some participants were at low risk for this adverse reaction. In a new study, the sponsor is trying to use an AI model to stratify patients, and participants with low risk for the adverse reaction will be sent home for outpatient monitoring after dosing. For this example, the question of interest would be “Which participants can be considered low risk and do not need inpatient monitoring after dosing?”
-
Example 2 (commercial manufacturing): Drug B is a parenteral injectable drug dispensed in a multidose vial. The volume is a critical quality attribute for the release of vials of Drug B. A manufacturer is proposing to implement an AI-based visual analysis system to perform 100% automated assessment of the fill level in the vials, to enhance the performance of the visual analysis system and identify deviations. For this example, the question of interest would be “Do vials of Drug B meet established fill volume specifications?”
A variety of evidentiary sources may be used to answer the question of interest, including evidence generated from the AI model. These different evidentiary sources should be stated when describing the AI model’s COU in step 2 and are relevant when determining model influence as assessed in step 3.
B. Step 2: Define the Context of Use for the AI Model
The COU defines the specific role and scope of the AI model used to address a question of interest.
The description of the COU should describe in detail what will be modeled and how model outputs will be used. The COU should also include a statement on whether other information (e.g., animal or clinical studies) will be used in conjunction with the model output to answer the question of interest.
C. Step 3: Assess the AI Model Risk
Model risk is the possibility that the AI model output may lead to an incorrect decision that could result in an adverse outcome, and not risk intrinsic to the model.
The Guidance recommends to assess the model risk with two factors: model influence and decision consequence. Model influence is the contribution of the evidence derived from the AI model relative to other contributing evidence used to inform the question of interest. In the clinical development of Drug A example provided before, model influence would likely be estimated to be high because the AI model will be the sole determinant of which type of patient monitoring a participant shall undergo.
Decision consequence describes the significance of an adverse outcome resulting from an incorrect decision concerning the question of interest. In the clinical development example of Drug A, the decision consequence is also high because if a participant who requires inpatient monitoring is placed into the outpatient monitoring category, that participant could have a potentially life-threatening adverse reaction in a setting where the participant may not receive timely and proper treatment.
The assessment of model risk is the prerequisite for determining the implementation of varying levels of model risk management measures. The Guidance expects that sponsors will establish appropriate risk management measures through effective assessment and, when necessary, demonstrate to regulators both the scientific validity of their risk assessments and the degree to which risk management measures align with assessment findings. For example, the performance acceptance criteria should be more stringent and described to FDA in more detail for high-risk models compared to low-risk models.
D. Step 4: Develop a Plan to Establish AI Model Credibility Within the Context of Use
For sponsors and other interested parties, the core compliance task in AI governance is to establish a credibility assessment framework for AI models and ensure robust evaluation of the credibility of AI model outputs. The Guidance discusses general considerations and assessment activities related to establishing and evaluating the credibility of AI model outputs. These suggestions are not meant to be exhaustive and some may not be applicable for all AI models and contexts of use.
FDA indicates that whether, when, and where the plan will be submitted to FDA depends on how the sponsor engages with the Agency, and on the AI model and COU. FDA strongly encourages sponsors and other interested parties to engage early with FDA to discuss the AI model risk, the appropriate credibility assessment activities for the proposed model based on model risk and the COU. The proposed credibility assessment plan about which the sponsor engages with the Agency should, at a minimum, include the information described in steps 1, 2, and 3 (i.e., question of interest, COU, and model risk) and the proposed credibility assessment activities the sponsor plans to undertake based on the results of those steps.
The requirements for model description and development are detailed in Table 1, while those for model evaluation are detailed in Table 2.
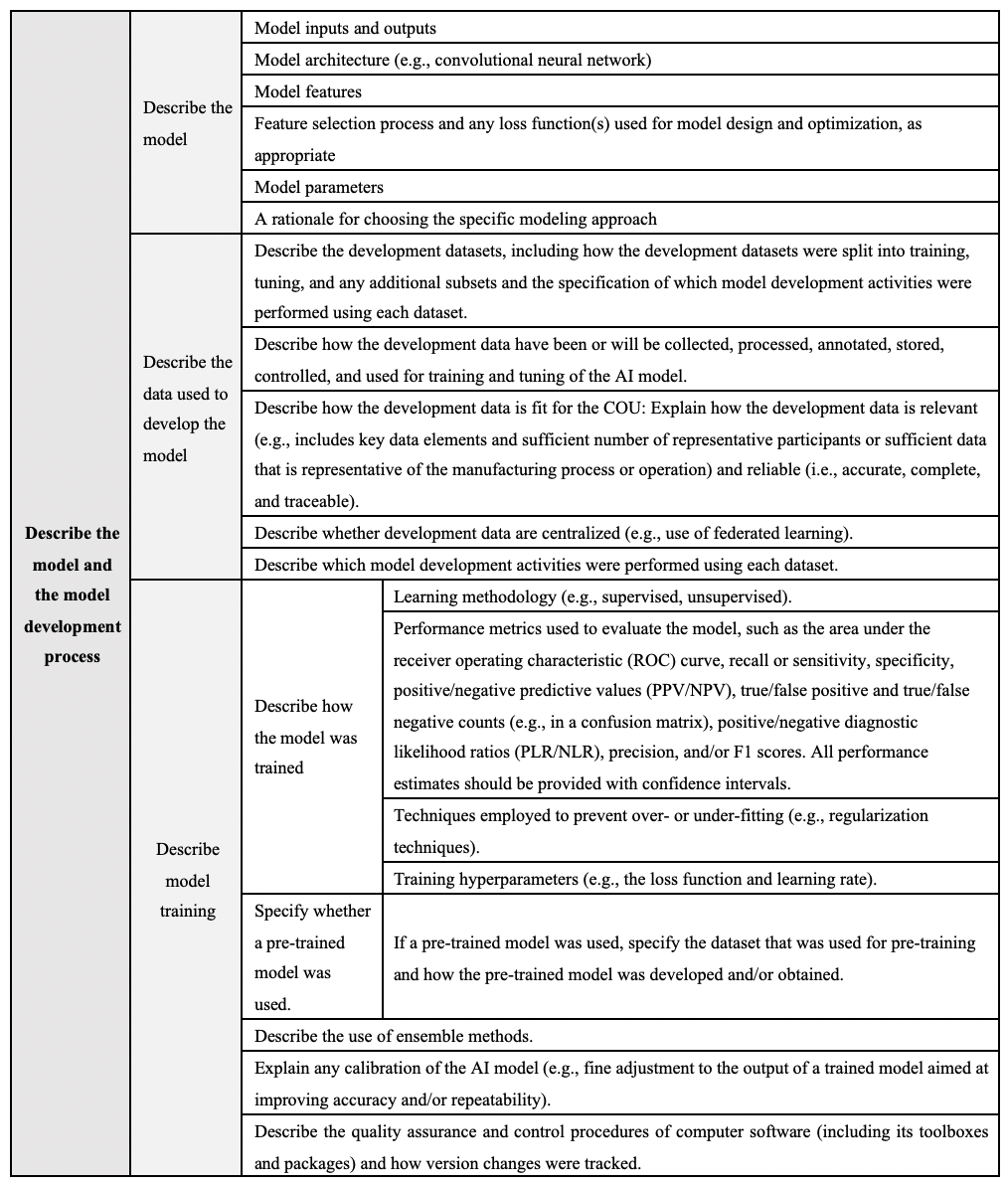
Table 1 Model description and development requirements
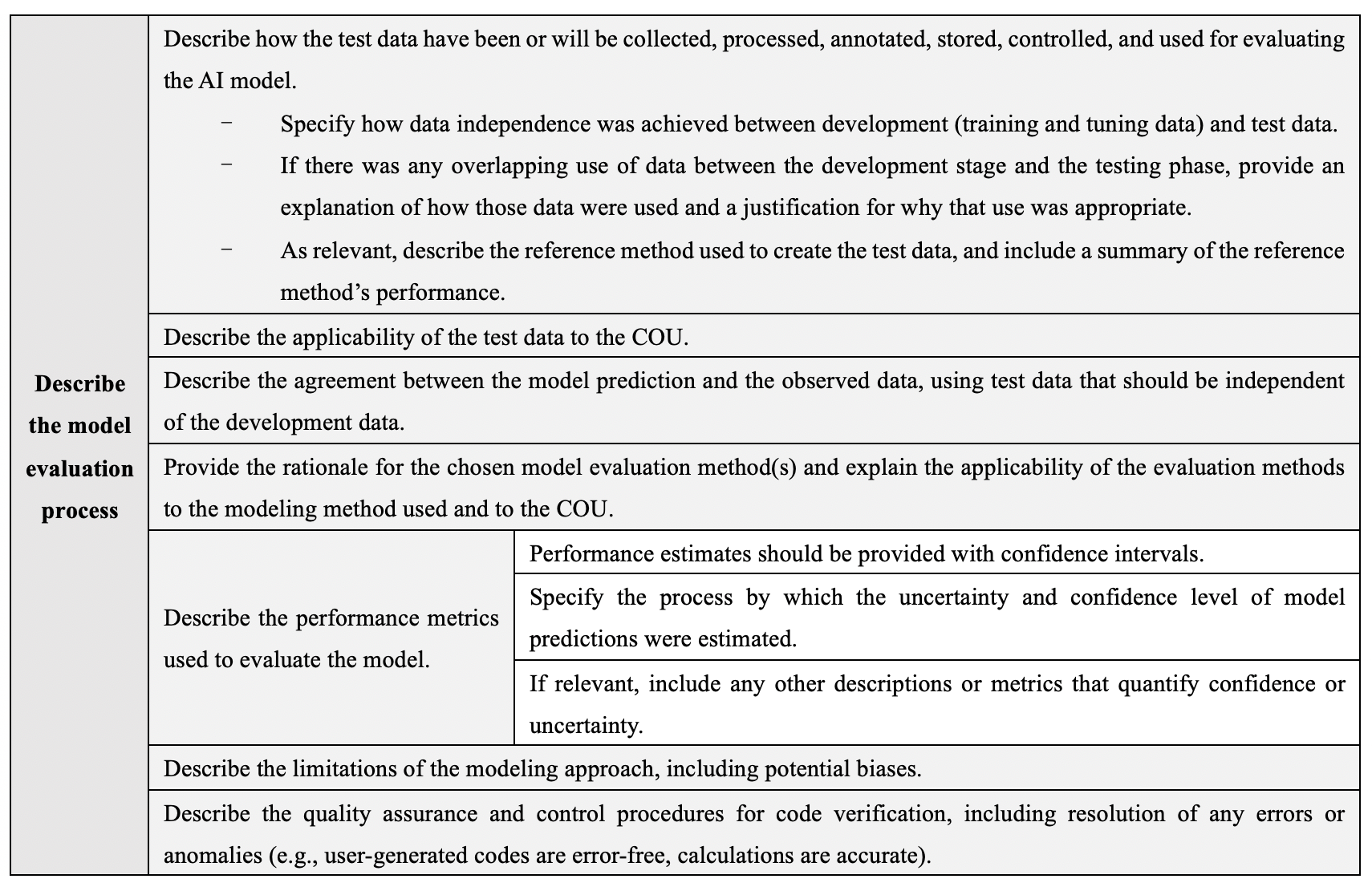
Table 2 Model evaluation requirements
E. Step 5: Execute the Plan
The Guidance encourages sponsors to discuss the plan with FDA prior to execution, in order to set expectations regarding the appropriate credibility assessment activities for the proposed model based on model risk and COU and to identify potential challenges and how such challenges can be addressed.
F. Step 6: Document the Results of the Credibility Assessment Plan and Discuss Deviations From the Plan
This step involves documenting the results of the credibility assessment plan and any deviations from the plan.
During early consultation with FDA, the sponsor should discuss with FDA whether, when, and where to submit the credibility assessment report to the Agency. The credibility assessment report may, as applicable, be (1) a self-contained document included as part of a regulatory submission or in a meeting package, depending on the engagement option, or (2) held and made available to FDA on request (e.g., during an inspection). Submission of the credibility assessment report should be discussed with FDA.
G. Step 7: Determine the Adequacy of the AI Model for the Context of Use
Based on the results documented in the credibility assessment report, a model may or may not be appropriate for the COU.
If either the sponsor or FDA determine that model credibility is not sufficiently established for the model risk, several outcomes are possible:
(1) the sponsor may downgrade the model influence by incorporating additional types of evidence in conjunction with the evidence from the AI model to answer the question of interest;
(2) the sponsor may increase the rigor of the credibility assessment activities or augment the model’s output by adding additional development data;
(3) the sponsor may establish appropriate controls to mitigate risk;
(4) the sponsor may change the modeling approach; or
(5) the sponsor may consider the credibility of the AI model’s output inadequate for the COU; therefore, the model’s COU would be rejected or revised.
IV. Public Comments on the Proposed AI Rules
As of April 2, 2025, the proposed Rules attracted 27 comments from the public[2]. Key themes are summarized below, reflecting the high complexity of the issue and the industry concerns over the compliance roadmaps and related ethical, regulatory or legal issues.
-
Current proposed model risk management measures are for traditional machine learning (ML) models, but not for Large Language Models (LLM). Model evaluation standards should be different for LLM than for ML models. The application of LLMs differs significantly from traditional language models.
-
Simpler explainable AI models may need loose requirements as compared with black box models. One company comments that its own AI is an explainable AI platform/white box, different from black box machine learning (ML) AI platforms. The Guidance should include non-ML AI platforms and set respective regulatory expectations and their assessments.
-
There was no discussion on different levels of model risks arising from model development process. The Guidance lacks specific recommendations in mitigating different forms of models risks such as bias, overfitting, interpretability, generalizability, ethical or regulatory.
-
Trade secret protection is also a key concern. On a few topics covering model transparency, interpretability, documentation, and regulatory submission, proprietary datasets and model code information are mentioned for confidentiality protection.
-
The Guidance does not explicitly discuss ethical and privacy considerations in AI models, including data security, algorithmic bias, and patient safety. Fairness metrics for different populations (e.g., race, gender, age, socioeconomic status) are suggested to identify disparities and to address unintentional biases. One company suggests FDA develop guidelines on how to report and mitigate such biases throughout the lifecycle of AI models.
-
The relationship between AI developers and AI deployers is requested to be included into consideration. Third party vendors may not want to share proprietary information on model training and valuations.
- Concerns are raised on the regulator’s capability of enforcing such AI governance rules. One question is how FDA will provide necessary training and expertise to enable its staff to evaluate AI models.
V. Recommendations for AI Governance in the Pharmaceutical Industry
The FDA’s Guidance is still in the public consultation stage. However, the FDA’s proposed requirements for data governance and model risk management are generally aligned with those of other jurisdictions, such as the EU and Japan. Based on the current trends in AI governance legislation and regulatory frameworks across various jurisdictions, it is highly likely that the main content of the Guidance would not undergo major changes. Uncertainty may arise from different policy priorities among countries and the potential shifts in US regulatory attitudes under the new administration.
For domestic pharmaceutical companies that are already applying or planning to use AI tools, it is crucial to consider the potential impacts of the new regulatory requirements when evaluate the time-consuming and cost of drug development, and the following key issues are recommend to take into consideration for industrial companies:
1. Comprehensive assessment
Companies should systematically review their AI models and assess potential risks to determine appropriate AI governance methods. Regulatory agencies like the FDA emphasize a “risk-based” governance approach. For lower-risk models or those where risks can be mitigated through alternative measures, companies may consider simpler AI governance to strike a balance between regulatory compliance costs and economic benefits.
2. Plan and manage AI development and application data
From the development phase onward, companies should establish standardized management practices for data collecting and sourcing. A thorough evaluation of data characteristics, potential data issues, and their impact on model development and testing should be conducted. Companies should implement standardized management processes, document identified issues and solutions, and establish mechanisms for validating, explaining, and tracing model performance and evaluations.
3. Closely monitor regulatory policy updates and prepare for compliance reporting in advance
When preparing for submissions to the FDA, companies should optimize workflows and documentation in accordance with data and model governance requirements, embed regulatory compliance measures into business and management processes early and enhance efficiency. Chinese companies involved in cross-border regulatory submissions should pay particular attention to data cross-border transfer compliance issues, proactively assess risks, and engage with domestic regulatory authorities as needed.
4. Maintain effective internal communication
From a compliance perspective, the pharmaceutical industry should establish strong internal collaboration between R&D units, AI development and application teams, and legal compliance teams. Effective interaction among technical, managerial, legal, and compliance domains is essential. When working with external AI developers, companies should emphasize collaboration strategies and communicate efficiently to facilitate the implementation of compliance requirements.
5. Engage in continuous communication with regulatory authorities
The Guidance highlights the importance of timely communication between sponsors and the Agency. Given the high technical complexity of AI governance and the diverse application scenarios, no single solution can address all challenges effectively. Companies should proactively engage with regulatory authorities to identify issues early, coordinate efficiently, and strive to reach consensus on challenges and solutions.
[1] https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-use-artificial-intelligence-support-regulatory-decision-making-drug-and-biological
[2] https://www.regulations.gov/docket/FDA-2024-D-4689/comments
