




2026年1月16日,国务院发布了第828号国务院令,公布了新修订的《中华人民共和国药品管理法实施条例》(以下简称《药品管理法实施条例》),《药品管理法实施条例》将于2026年5月15日起施行。这是对2002年公布施行的《药品管理法实施条例》的首次全面修订。与23年前相比,我国药品研制、生产、流通、使用各环节都发生巨大变化,法律法规制度也需要顺应变化,修订完善。
此次条例修订,明确支持以临床价值为导向,研究和创制新药,并首次引入了儿童用药品、罕见病药品市场独占期制度和数据保护制度。
市场独占期和数据保护期制度的引入对于中国药品的监管具有里程碑意义:这使得中国市场上的创新药,在专利以外,还可以获得新的制度保护,市场独占期制度和数据保护制度提供的制度性红利将进一步赋能创新主体更好地服务“健康中国”的国家战略。
虽然,市场独占期制度和数据保护制度对于中国创新主体而言是新事物,但在美国和欧洲,前述两个制度已经运行多年。本文拟以中、美、欧三大代表性药品市场的药品保护期制度为研究对象,系统比较其专利排他期(如专利期限补偿)与监管排他期(如试验数据保护、市场独占期)的立法模式与实践操作,剖析其制度差异与运行效果,并最终为我国本土药企在“出海”过程中如何应对不同司法辖区的药品保护制度提供有针对性的法律策略和制度应对路径。
一、概念辨析[1]
厘清基本概念是任何研究的起点。在讨论中美欧药品保护期的差别之前,下述基本概念应该先行确定,并对其内涵进行阐述。
(一)基石概念
专利排他期(Patent Exclusivity, PE):专利排他期是指药品作为一种技术方案获得专利权后,其专利权人在法定期间内(对于发明专利而言,通常为自申请日起20年)享有排除他人实施该专利的权利。值得注意的是,针对药品领域,中美欧均特别设置了药品专利期补偿制度,如美国的专利期限延长(Patent Term Extension, PTE)、欧盟的补充保护证书(Supplementary Protection Certificate, SPC),中国《专利法》第四十二条第三款[2]也规定了相应的补偿机制。对此,下文将展开详细介绍。
数据排他期(Data Exclusivity, DE):数据排他期是指原研药企基于临床试验数据等数据(上市许可数据)提交新药申请并获批上市后,其他人不得利用或参照该许可数据获得上市许可。该制度的国际法律基础在于《TRIPS协定》第39.3条:“成员应保护提交以获得药品营销许可所需的新化学成分的、通过相当努力取得的未披露的试验数据不被不正当商业使用。”该项规定对于含有新化学成分的药品,为获得药品批准而向药品注册部门提交的,能够证明药品安全性和有效性的未披露的试验数据,进行了保护。美国《联邦食品、药品与化妆品法》(FDCA)第505条、欧盟2001/83/EC第10条、《药品管理法实施条例》第22条均设有相应制度,但数据排他期根据药品的种类以及法域的不同,会有较大差异,下文将进行详细讨论。
市场排他期(Market Exclusivity, ME):市场排他期是指在一定期限内,仿制药,生物类似药等不得进入市场进行商业销售的时间限制,无论该等竞争产品是否已经获得上市许可。市场排他期效力强于数据排他期,因为即便竞争企业独立完成试验并获得批准,也不得销售。类似于数据排他期,市场排他期也随着药品种类和法域的区别,有较大差异,下文将进行详细讨论。
(二)基石概念的异同比较
上述三种排他期的共性在于:均在一定期间内限制竞争性产品进入市场,从而鼓励创新。美国食品药品监督管理局(FDA)官网对于Exclusivity有更为精准的定义:排他性是指在药品或某些补充药品获得批准后,根据法规对竞争药品的批准附加的某些延迟和禁令。[3]
但虽同为排他期,三者在适用路径、保护范围、权利证明与实施机制等方面仍存在显著区别。
首先,数据排他期和市场排他期均属于监管独占期项下的内容(Regulatory Exclusivity)。不同于专利保护期,监管独占期是药品领域中特有的一种非专利法律保护制度,其目的在于通过监管法律赋予原研药企在一定时间内不受仿制药或竞争产品进入市场的“排他性利益”,即使该原研药没有专利或其专利已到期,仍可在排他期内阻止仿制药通过依赖其注册数据或直接上市而竞争。数据排他期和市场排他期是监管独占期的两种主要形式。通过数据排他期,仿制药公司在一定时间内不得依赖上市许可数据来支持其相同药品相同适应症的上市许可申请,但数据排他期并不能阻止其他公司提交自己的数据信息并获得上市许可申请。相比之下,市场排他期的效力则更为绝对,市场排他期直接会阻止竞争公司进入市场,无论该竞争产品是否生成了自己的安全性和有效性验证,亦无论其是否取得了药品监管部门的批准。
下表总结三种排他期主要差别比较(非穷尽):
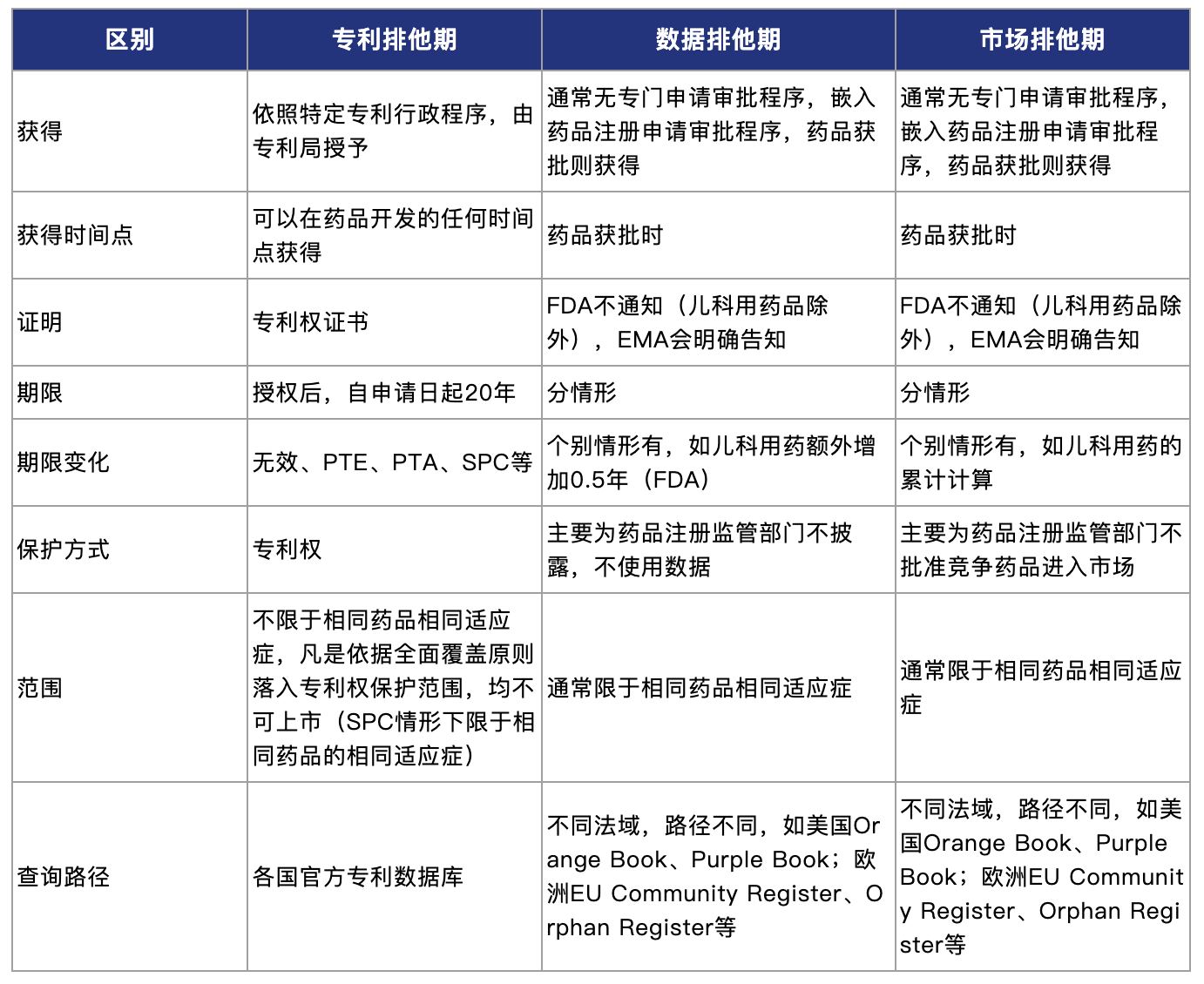
需要指出的是,专利排他期、数据排他期、市场排他期市是独立运行的法律制度,彼此之间并无绝对的关联关系。一个药品可以同时享有三种排他期,也可能只享有其中一种或两种。专利排他期长于、短于或等于数据或市场排他期均属于正常情况。
(三)药品试验数据不被披露与不被依赖
在药品实验数据保护制度下,保护的实施者为政府机构。为获得药品批准上市,制药企业按照法律要求将药品试验数据递交给监管机构,政府机构虽然掌握企业递交的数据,但不能随意处置和利用申请人提交的试验数据,政府有义务保护试验数据不被披露与不被依赖。
“防止披露”相对易于理解,即政府机构不得向未经授权的第三方公开或泄露企业提交的试验数据。然而,该条款亦保留了例外情形:为维护公众健康或知情权,监管机构可在保障数据不被不正当商业使用的前提下,适度披露部分信息。例如,FDA可披露部分药品的安全性数据,以利公众知情。
相较之下,“防止依赖”值得进一步阐释,目前大多数发达国家,包括美国和欧盟对数据保护均采取数据专属权模式[4],该模式强调在数据保护期内,任何人不得使用或依赖原始数据,包括政府不得基于这些数据批准仿制药上市。需要明确的是,并非所有制定药品实验数据保护的国家都采用此种模式,部分发展中国家,例如阿根廷、巴西、南非、印度等采用的是禁止占用(商业秘密)模式,在该模式下,政府机构可依据制药企业递交的药品数据来审批仿制药的上市申请,本质上该模式有利于仿制药尽快入市。
“依赖”(rely on)最早出现在《北美自由贸易协定》(NAFTA)中,NAFTA第1711.6条规定缔约方应规定在申请人提交上市许可申请后一段合理期间内,除提交数据的申请人以外,他人不得依赖受保护数据支持后续产品的上市许可申请。条约赋予原创药品持有人试验数据一定期限市场独占保护的权利,但是没有解释何种行为构成“依赖”,NAFTA的缔约国加拿大曾在本国的《食品和药品法规》(The Food and Drug Regulations)中规定了五年的药品数据保护期并在相关规定中照用了“依赖(rely on)”一词[5],但对其作出了狭窄的司法解释。在1998年的拜耳诉加拿大卫生部长案(Bayer Inc. v. Canada (Minister of Health), Federal Court of Canada, 84 C.P.R. (3d) 129)中,原告拜耳公司向负责药品监管审批的部长提交了一份新药申请并认为应按照C.08.004.1获得五年数据保护期。拜耳公司认为,卫生部部长在通过简化仿制药流程(ANDS)[6]批准与原研药生物等效的仿制药时,不可避免地会通过原研药企业提供的已经证实药品安全性和有效性的信息(即“间接依赖”),因为监管机构审查仿制药制造商的提交时,只需证明其产品是原创产品的药学和生物等效物,而无需审查等效药的安全性、有效性和质量,因为这些标准已通过原创者的临床数据确定。加拿大法院最终裁定:只有在监管机构实际查阅并依赖了创新药的数据时,数据保护期才适用;若审批过程中未直接依赖该数据,则数据保护机制不适用。法院明确指出,“rely on”必须是“实质性查阅”,间接参照并不足以触发数据独占期。
该判决被批评为过于狭隘,未能充分保护原始数据。为回应这一问题,加拿大于2006年再次修订《食品与药品法规》,明确规定政府机构在八年的数据独占保护期间内不得批准任何直接或间接与原创药进行对比基础上提交的仿制药申请。数据保护不再以监管机构“依赖”为前提,而是转变为政府机构在一定期限内不得审批和(或)批准依赖原创药数据的仿制药上市,除非仿制药企业自己通过临床试验提交NDA或者BLA而非依赖原研药企业的数据,通过制度上的不受理和不审批来实施对试验数据不被依赖的保护。
二、中美欧涉药品保护期的相关制度比较研究
(一)中国
药品研发具有高投入、高风险和研发周期长的特点,当赋予药企一定时期的创新药物市场独占权,使其能够通过高价回收研发成本并盈利,有助于激励企业持续投入创新。伴随着我国医药创新生态体系的不断完善,为鼓励和保护药品创新,药品专利保护、专利期限补偿、药品专利保护期限补偿制度、药品试验数据保护制度、药品市场保护制度等行政保护制度也日臻完善。
1. 中国药品专利排他保护制度
1984年我国颁布的首部《中华人民共和国专利法》中对药品的制备方法予以保护,但第二十五条对药品和用化学方法获得的物质不授予专利权,1992年修订的《专利法》取消了对药品和化学物质专利的排除条款,自此药品获得了20年的全面专利保护。2020年修订的《专利法》新增了专利权期限补偿制度和药品专利保护期限补偿制度。2023年修订的《中华人民共和国专利法实施细则》第五章和《专利审查指南(2023)》对专利权期限补偿进行了进一步的细化和补充。
(1)专利权期限补偿
专利权期限补偿制度是对因国务院专利行政部门在专利授权过程中造成的不合理的期限延误进行补偿,其补偿范围涵盖了包括药品发明专利申请在内的全部发明专利,有助于延长了药品发明专利的价值期限。2020年修订的《专利法》第四十二条第二款规定:“自发明专利申请日起满四年,且自实质审查请求之日起满三年后授予发明专利权的,国务院专利行政部门应专利权人的请求,就发明专利在授权过程中的不合理延迟给予专利权期限补偿,但由申请人引起的不合理延迟除外。”其中专利权人需要在专利被授权之日起三个月内向授予专利权的行政部门提出补偿申请,且专利期限补偿请求不适用时间期限的恢复申请,因此,专利权人应当注意按时提出专利权期限补偿请求。专利权期限补偿天数=总延迟天数-合理延迟天数-申请人引起的不合理延迟天数,其中总延迟天数为自发明专利申请日起满四年且自实质审查请求之日起满三年之日至公告授予专利权之日的间隔天数。合理延迟的情形包括:修改专利申请文件后被授予专利权的复审程序、行政诉讼程序、中止程序、保全程序以及其他合理情形引起的延迟。由申请人引起的不合理延长则包括:未及时答复审查意见通知书导致的延迟、申请延迟审查导致的延迟、援引加入补交申请文件导致的延迟、自优先权日起30个月内办理进入中国国家阶段手续的国际申请,申请人未要求提前处理引起的延迟。
(2)药品专利权期限补偿(≤5年)
药品在上市之前,需要经历药物发现、非临床研究、临床研究、药品审批等多个阶段,该过程耗时长、投入高,且医药企业无法从新药中获得直接经济利益,而当在研药品显示出临床潜力时,为了获得专利保护,医药企业往往在药物发现或非临床研究阶段就提交专利申请,而药品上市审评用时较长,综合导致了药品获得上市批准后药品剩余专利保护期相应较短,专利到期后仿制药以其研发周期短、研发投入少等优势,迅速挤占原研药企的市场份额,影响原研药企的实际收益,降低研发新药的热情。2017年10月,国务院印发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,明确表示要“开展药品专利期限补偿制度试点”。2020年修订的《专利法》第四十二条第三款正式规定了药品专利期限补偿制度,药品专利权期限补偿=(中国上市许可日-专利申请日)-5年,其中最长补偿五年,且要求自新药上市日起算,总有效专利权期限不超过十四年。需要注意的是,这里的新药仅包括人用药物,并不包含兽药农药,也不包括医疗器械,且补偿对象仅限于新药相关发明专利,不包括实用新型,也不包括外观设计。
药品专利权期限补偿的提出同样要求专利权人自该新药在中国获得上市许可之日起3个月内向国务院专利行政部门提出,且要求该专利在有效期内,其尚未获得过新药相关发明专利权期限补偿。当该新药同时存在多项专利时,只能对其中一项专利给予专利期限补偿,而当一项专利同时涉及多个新药的,只能对一个新药就该专利提出专利权期限补偿请求。依据《专利审查指南(2023)》,“新药”是指针对国务院药品监督管理部门批准上市的创新药和符合规定的改良型新药,对于上述新药相关的药物活性物质的产品发明专利、制备方法发明专利或者医药用途发明专利,可以给予药品专利权期限补偿。改良型新药则包括了:化学药品第2.1类中对已知活性成分成酯或成盐的药品、化学药品第2.4类含有已知活性成分的新适应症的药品、预防用生物制品第2.2类中对疫苗菌毒种改进的疫苗、治疗用生物制品第2.2类中增加新适应症的生物制品,以及中药第2.3类增加功能主治的中药。
药品专利权期限补偿的目的是补偿新药审批上市所导致的药品专利保护期限的缩短,如果药品上市许可时间早于专利授权时间,即药品上市许可并未导致药品专利保护期限的缩短,则失去了进行相关药品专利权期限补偿的必要性,因此,《审查指南》第五部分第九章3.1节明确了请求专利补偿的专利授权公告日应当早于上市许可申请获得审批之日。另外,药品期限补偿期间内,该专利的保护范围限于国务院药品监督管理部门批准上市的新药,且限于该新药经批准的适应症相关技术方案,因此,药品专利权期限补偿的范围可能小于相应专利的保护范围。
2. 中国药品监管排他期保护制度
(1)药品数据排他期保护制度(≤6年)
新颁布的《药品管理法实施条例》第二十二条规定:国家对含有新型化学成份的药品以及符合条件的其他药品的上市许可持有人提交的自行取得且未披露的试验数据和其他数据实施保护,任何人不得对该未披露的试验数据和其他数据进行不正当的商业利用。
前款规定的数据的保护期限自药品注册之日起不超过6年。在保护期限内,其他申请人未经药品上市许可持有人同意,使用前款规定的数据申请药品注册的,不予许可;但是,其他申请人提交自行取得数据的除外。
除下列情形外,药品监督管理部门不得披露本条第一款规定的数据:
(一)公共利益需要;
(二)已采取措施确保该类数据不会被不正当地进行商业利用。
我们期待配套的实施细则对“不正当的商业利用”和“不超过6年”的细化定义。在细化定义出台前,我们可以先从2025年3月19日国家药品监督管理局发布的《药品试验数据保护实施办法》(征求意见稿)中寻找一些可能的提示:
-
对含有新型化学成份的药品以及符合条件的其他药品获批上市时,对申请人提交的自行取得且未披露的试验数据和其他数据实施保护,给予最长不超过6年的数据保护期。在数据保护期内,其他申请人提交自行取得数据申报药品注册申请的,符合条件的予以批准,不再给予数据保护期,但该数据不得被后续其他申请人依赖。
-
自首次境内上市许可之日起,创新药给予6年数据保护期,改良型新药给予3年数据保护期,境外已上市境内未上市的原研药品申请在境内上市的,数据保护期限为6年减去该药品在境内提交上市许可申请被受理之日与该药品境外首次获得上市许可之日的时间差。境外已上市境内未上市的改良型药品申请在境内上市的,数据保护期限为3年减去该药品在境内提交上市许可申请被受理之日与该药品境外首次获得上市许可之日的时间差。
-
对首家获得批准的境外已上市境内未上市原研药品的仿制药(含境外生产药品)和生物制品给予3年数据保护期。
该版征求意见稿按照创新药、改良型新药、仿制药和生物制品对数据保护期进行区分,调整了数据保护期的计算机制,涵盖了境外与境内首次上市时间差的扣减规则、首仿激励等内容,旨在促进药品创新和仿制药发展,完善药品试验数据保护制度,对申请人提交的自行取得且未披露的试验数据和其他数据进行分类保护。
我们期待并持续关注后续正式发布的配套实施细则的具体规定。
(2)药品市场排他期保护制度(儿童用药品≤2年,罕见病药品≤7年)
新颁布《药品管理法实施条例》第二十一条规定:
“国家支持儿童用药品、罕见病治疗用药品的研制和创新。
对儿童用药品新品种、采用新剂型或者新规格的儿童用药品、增加儿童适应症的药品,符合条件的,给予不超过2年的市场独占期。
对符合条件的罕见病治疗用药品,药品上市许可持有人承诺保障药品供应的,给予不超过7年的市场独占期。药品上市许可持有人不履行保障药品供应承诺的,市场独占期终止。
给予市场独占期的具体条件和办法,由国务院药品监督管理部门制定。”
市场排他期制度首次被引入中国药品监管体系,条文本身即反映了监管层对儿童用药品和孤儿药的政策倾斜。对于儿童用药品,新剂型、新规格、新适应症都将被政策红利覆盖,这对以儿科制药为强项的企业而言,是一个巩固市场优势地位的契机。
我们期待并持续关注后续的配套实施细则对儿童用药品的市场排他期是否能够叠加、是否要求临床试验成功等作出的更细节的规定。
(二)美国
在美国,药品保护主要通过两种制度实现:专利保护和监管排他期保护,其中监管排他期保护具体包括数据排他保护期和市场排他保护期。
1. 美国药品专利排他保护制度
美国在针对药品提供20年专利保护的基础上,还制定了专利保护期延长制度和专利权期限调整制度,分别用于补偿因药品上市的行政审批、专利审查所导致的药品专利保护期的损失。
(1)专利期补偿(Patent Term Extension,PTE)
PTE起源于美国1984年的《药品价格竞争与专利期补偿法案》(PL 98-417、S1538 98 Stat.1585)(即Hatch-Waxman法案)。通常,美国专利的有效期自提交专利申请之日起20年,由于药品和设备在商业使用或推出之前必须经过FDA的漫长审查过程,而相关专利通常在FDA批准之前很久就已开始提交(从而开始20年的期限),因此Hatch-Waxman法案第二章,即专利期限恢复或专利期限延长部分,通过专利延长制度为药品专利持有人(包括抗生素和生物制剂、医疗器械、食品添加剂和颜色添加剂)提供了延长专利期限的机会,以补偿因联邦监管审查而失去的部分专利期限。
可以提出专利期限延长的专利必须要求专利期限尚未到期,专利保护产品、使用产品的方法或制造产品的方法,且专利此前未被延长过,目前尚未有其他任何专利期限因该产品的监管审查期而延长。即一件专利只能与一个产品相绑定,不存在一个产品多件专利获得保护期延长或者一件专利因多个产品获批延长。其中“产品”包括药品(包括新药、抗生素药品、人类生物制品或者新动物药品或兽医生物制品,且主要不是使用重组DNA、重组RNA、杂交瘤技术或其他涉及特定基因操作技术的工艺制造的),以及任何根据《联邦食品、药品和化妆品法》受到监管的医疗设备、食品添加剂或颜色添加剂。
申请必须在产品获得FDA商业营销或使用许可之日起60天向美国专利商标局提交申请。如果错过截止日期,专利持有人可能会失去 PTE 权利。
美国药品专利期限补偿时间PTE=(NDA获批的总时间-专利授权日前NDA审批的时间-专利权人未尽到合理注意义务延误的时间)+(临床试验的总时间-专利授权日前临床试验的时间)×½,即美国药品专利期限补偿时间PTE≈½(临床试验阶段)+批准阶段。
(2)专利权期限调整制度(Patent Term Adjustment, PTA)
PTA用于补偿因专利授权审查流程导致的延误时间。根据35 U.S.C.154(b),美国专利商标局(USPTO)审查过程可以包括三种延迟类型,即A类延误、B类延误和C类延误。A类延误是指USPTO未在规定期限内发出相应的通知书或做出回应,B类延误是指USPTO未在专利申请实际提交日起3年内颁发专利做出授权决定,C类延误是指因派生诉讼、保密令或上诉程序且经审查推翻了对可专利性的不利决定的裁决而授予专利。美国专利期限调整计算方式可表示为PTA=A类延误+B类延误+C类延误-三类延误重叠时间-因申请人原因延误的天数。
2. 美国药品监管排他期保护制度
监管排他期保护通常指的是新药上市后,FDA给予原研药厂一定期限的市场独占权,以激励创新药物的研发。这种排他性可以阻止仿制药或生物类似药在一定时间内上市,确保原研药厂能够回收研发投入并获得合理的回报。
(1)新化学实体临床数据保护期(5年)
新化学实体(New Chemical Entity,简称NCE)是指以前没有被用于治疗疾病的药物产品。它不包括现存药物的新型盐类、前药、代谢物和酯类等,也不包括已知药物的组合物。根据美国FDA的规定,NCE必须含有之前未被FDA批准的活性成分,它不含已依据《食品、药品和化妆品法案》提交申请或已在美国上市的药品的活性实体。
NCE临床数据保护期源于1984年9月24日通过的《药品价格竞争和专利期恢复法》(Hatch-Waxman法案),该法案中首先引入了一种新型的仿制药市场批准申请——“简化新药申请”(ANDA)的概念,即如果仿制药的活性成分与已批准药物的生物等效性相同,则可以提交ANDA。ANDA允许仿制药制造商依赖原始制造商的安全性和有效性数据,而不必承担与提交完整的新药申请(NDA)相关的成本和延误。大多数情况下,ANDA允许仿制药在相关专利到期后立即上市,除非原研药厂享有数据保护期或市场独占期。
在便捷仿制药制造商的同时,该法案通过引入数据保护期来对仿制药竞争者引用创新药制造商生成的数据的能力施加一定的限制。Hatch-Waxman法案给予新化学实体(NCE)5年的数据保护期,从NDA获得批准之日起计算。在NCE排他性的五年期间,FDA不得接受仿制药公司提出的药物上市申请,即销售包含受NCE排他性保护的相同活性部分的药品。即使这些应用针对活性成分的不同用途、剂型或酯或盐,此禁令也适用。因此,如果FDA花了2年时间审批,则仿制药在7年内都无法通过ANDA上市。
然而,如果仿制药申请包含所谓的Paragraph IV Certification,即仿制药申请人认为原研药的专利无效或不会因仿制药的上市而侵权,则数据保护期将减少到4年。在这种情况下,仿制药申请人可以在NDA获得批准后4年届满时提交申请,而不是5年。第一个向美国FDA递交ANDA、并含有PIV声明的仿制药申请者,如果专利挑战成功,则FDA将给予180天的市场独占期。在这180天内,FDA不再批准其他的ANDA持有人上市。通过专利挑战激励与市场竞争促进的双重机制,在保护原研药创新与提升仿制药可及性之间取得平衡。
(2)生物药市场排他期(12年)
《生物制剂价格竞争和创新法案》(BPCIA)作为《患者保护和平价医疗法案》的一部分颁布,该法案为“生物制剂”(biologics)引入了新的监管排他性。根据42 U.S.C第262条,生物制剂包括疫苗、抗毒素、血液成分和治疗性血清等产品。
BPCIA修订了《公共卫生服务法》(PHS Act)和其他法规,为证明与FDA许可的生物仿制药与原研药具有生物相似性或可互换性(见《平价医疗法案》第7001至7003条)。
PHS法案第351条(k)款第(7)项为“原研药品的排他期”,即自原研药品首次获得上市许可的一定期限内仿制药企不得提交生物类似药申请,而FDA也不得参照原研药品批准生物类似药申请。具体而言,自原研药品按照PHS法案第351条(a)款首次获批上市起4年内 FDA 不得接受任何基于该产品数据的生物类似药(biosimilar)或可互换产品(interchangeable)的申请,自原研药品按照PHS法案第351条(a)款首次获批上市起12年内不得批准仿制药根据第351条(k)款提交的上市申请。
按照PHS法案第351条(m)款的规定,如果原研药品进行了儿科药物研究,其可以获得额外6个月的排他期(分别与4年和12年累计,即4.5年不得提交,12.5年不得批准)。此外,如果原研药品根据《联邦食品、药品和化妆品法》第527条(a)款享有7年的孤儿药排他期,那么,12年的数据排他期和7年的孤儿药排他期两者的在后终止日期为FDA可以批准生物类似药的起始时间。
(3)新临床试验保护期(New Clinical Investigation Exclusivity,3年)
对于已经获批上市的药品,如申办方就包含已上市药品活性成分的新剂型、新用途、新给药途径等进行了新的临床试验,申请或补充材料中包含由申请人进行或赞助的新临床研究(非生物利用度研究)报告,且这些研究对于获得批准至关重要,则已上市药品和在后有效申请的药品均可获得3年的新临床试验保护期。
(4)孤儿药市场排他期(7年)和儿科用药市场排他期(0.5年)
1983年,美国国会颁布了《孤儿药法案》,孤儿药是指罕见疾病和病症的药物,根据21 U.S.C.第360条规定,孤儿药是指(1)影响美国不到200,000人的罕见疾病或病症,或(2)影响美国超过200,000人,但没有合理期望该药物的销售能够收回成本。
为促进孤儿药的发展,FDA给予了一系列保护计划,其中就有7年的保护期,从FDA颁发上市许可之日起计算。该7年的保护期为市场独占期,也就是说,无论竞争对手是否生成了自己的数据,都无法获得FDA批准。孤儿药7年保护期仅适用于该药物获得批准的适应症。因此,FDA可以批准同一药物的用于不同的用途的二次申请。
与孤儿药类同,美国对儿科用药也给予了特殊的保护。根据21 U.S.C第355条,医药公司在完成关于药物对儿童影响的研究后,有机会获得6个月独占期,从创新药的现有专利或数据独占保护到期之日开始计算。并且该保护期延伸到具有相同活性成分(也称为药物的“活性部分”)的任何药品。值得注意的是,《食品和药物法》并未以研究成功为条件提供儿科排他性保护。
实践中,只要申办方“fairly ”回应了FDA的书面要求,并进行了研究,无论儿科适应症是否获批,均可获得PED。即,无论该研究是否成功证明在儿童中安全性和有效性,都可以获得六个月的监管排他期。儿科6个月排他保护期从现有的排他期或专利保护期计算,再延长六个月。
儿科排他期不单独存在,且PED是唯一可以直接在专利排他期到期后直接叠加的排他期(但其并不是专利排他期的延长),且可以与任何排他期直接叠加。6个月的儿科专属期不能从标签中“carved out”。当儿科用药排他期生效时,在桔皮书的专利栏中,该专利会显示两次——一次是原专利到期日,第二次是与该特定专利相关的六个月儿科排他期。
ANDA申请人在儿科排他期内无法获得ANDA申请的全面批准,除非ANDA申请人获得了儿科排他期权利人的豁免,或法院生效裁决确定叠加了儿科排他期的专利无效、未被侵犯或不可执行等。
例如,如果儿科药品排他性适用于孤儿药,结果将是7年零6个月的排他期保护;如果应用于NCE排他性,该药物的申办者将获得五年零六个月的数据保护。但值得注意的是,儿科药品排他期实际上并不会延长专利的期限,因为他是属于由FDA监管的排他性保护期的一种。
(5)抗生素药品市场排他期(Generating Antibiotic Incentives Now Exclusivity,GAIN ,5年)
一些新的抗生素针对特定感染有可能获得5年的排他期。GAIN可以和任何既存的排他期累计计算。2012年生效的Generating Antibiotic Incentives Now Act (GAIN Act)针对符合以下条件的“用于人类的抗菌或抗真菌药物”给予额外的5年市场保护期。药品是针对:对抗生素或抗真菌剂产生抗药性的病原体,包括新型或新出现的感染性病原体;《食品、药品和化妆品法》第505E条(f)款列出的病原体。
如:

GAIN可与NCE累计叠加计算。对于获得了GAIN药物的首次申请或补充申请,FDA将优先审查,申请人还可申请快速审查通道。
(三)欧洲药品排他保护制度
1. 欧洲药品专利排他保护制度
欧盟目前针对药品提供20年的专利保护期,并通过补充保护证书制度补偿上市审批导致的专利保护期的损失,另外,还配套了补充保护证书延长期内针对以出口为目的的生产、储存仿制药的豁免政策。
(1)补充保护证书(SPC,Supplementary Protection Certificate)制度
SPC由欧洲议会于1992年颁布通过((EEC)1768/92)[7],并于1993年生效,随后又修订为第EC/469/2009号条例,并于2009年在整个欧盟生效,2019年通过第2019/933号条例引入了“生产豁免”和“贮存豁免”,于2019年7月1日起正式生效。SPC制度是一项旨在补偿药品专利权人在寻求上市行政许可中损失的专利保护期而实施的专利延长保护期的制度,目的在于确保药品开发商收回研发投资并实现利益最大化。
SPC的保护对象为医药产品,且其内涵相比药事法中的内涵狭窄,第EC/469/2009号条例第一条规定:药物产品是指用于预防、治疗、诊断、恢复、矫正人体或动物体疾病,改善生理机能方面的物质和物质组合物。实践中,对能够授予药品补充保护证书的医药产品作了更为严格且狭义的解释,要求医药产品必须是有重大创新及拥有疗效的有效成分。此外,对于组合物而言,要求组合物中的有效成分具有独立性,并且各自具备疗效。对于赋形剂、辅料、新的剂型或者已知活性成分的新适应证通常无法获得专利保护期限的补偿,然而,例外情况也是有可能的,即当后续研发的产品(可能与另一种物质组合)提供新的治疗效果,即与早期SPC产品/早期专利的产品不等同/不相同时,并且其产品本身就从属于一项权利要求的情况下,获得基本专利之外的SPC是有可能的。
医药产品获得SPC需要满足四大条件(SPC法规第3条):(a)该产品受有效的基本专利保护;(b)该产品已获得作为药物产品在欧洲上市的有效许可;(c)该产品尚未成为证书的保护对象;(d)上述(b)点中提到的许可是该产品作为药品的首次上市许可。
一个SPC仅可用于一个产品(即如第1条所定义的获得上市许可的“产品”,但其中也包括活性成分的衍生物(盐,酯),如果该衍生物也包含在基本专利的保护中),并且一个SPC仅可用于一项专利。
欧盟药品补充保护的期限为药品上市日与药品专利申请日(专利期起算日)之间的时间差(专利折损期)再减去5年,即“保护期=专利折损期-5年”。同时药品补充保护证书的权利期限最长不超过5年(期限自专利失效日起算),而且因专利及药品补充保护证书所获得的市场独占权总期限不能超过15年(期限自药品上市日计算),对专利折损期少于等于5年的不予以补充保护。
此后Regulation(EC) No 1901/2006条例确立了SPC儿科规则,并于2007年1月26日正式生效。根据SPC儿科规则,已经获得药品补充保护证书的药品在满足额外条件时保护期限可以延长一次,延长期限为6个月。具体条件如下:首先,医药产品在所有欧盟成员国获得了上市许可;其次,按照议定的儿科临床研究计划(Pediatric Investigation Plan)进行的所有研究(步骤、措施等)符合要求;最后,申请文件包括根据议定的儿科临床研究计划所开展的所有研究的结果及详细资料。由此看来,欧盟药品补充保护证书制度的延长期限适用于那些开展了儿科临床试验研究的药品,其目的在于对儿科药研究投入的肯定,这种鼓励聚焦于对儿科药研究的努力与付出,与研究结果的成败无关。实际中,在所有欧盟成员国获得医药产品的上市许可并非易事,因而保护期的延长并不容易。
欧盟药品补充保护证书制度与专利制度相关联但却独立于专利制度,它不是对专利保护期限的简单延长,SPC只保护获得上市许可产品范围内相应的药品,以及该产品在SPC到期前已被授权药品的用途,无法保护在基本专利权利要求的全部保护范围。
第2019/933号条例在原第469/2009号条例的第5条“证书的效力”特别增加了“生产豁免”以及“贮存豁免”的例外规定以及对该例外规定的限制条件。如果在SPC保护到期之前的生产是为了将所生产的药品出口到欧盟之外的第三国或者为了在SPC保护到期之后马上进入欧盟市场,则该生产或者贮存行为不构成侵权,其中为了出口至第三国的,“生产豁免”适用于在SPC保护的全部期限内,如果生产受保护的活性药物成分或者包含该成分的药品是为了贮存并为SPC过期之后立即进入欧盟市场做准备的话,“生产和储存豁免” 适用于在不超过SPC过期之前的6个月内。需要注意的是,这两种“豁免”仅仅是SPC侵权的例外,如果被豁免的行为同时落入其它专利权的保护范围,那么该生产药品的行为仍然会构成专利侵权。
2. 欧洲药品监管排他保护制度
(1)药品实验数据排他期保护制度(Data Exclusivity,DE ,8年)
1987年,当时还称为“欧洲经济共同体”的欧盟通过了欧洲经济共同体理事会第87/21/EEC号条例,在该指令中首次引入了试验数据保护制度。在数据保护期内,药品主管部门不能够依赖原研药的安全性和有效性数据批准仿制药上市。随后经过2001和2004年两次修订,最终形成了数据保护的“8+2+1”方案。即药品试验数据保护期为10年,但在后2年里也就是新药上市8年后,仿制药生产商可以提交仿制药上市申请。如果新药在批准上市后的8年间增加了新的适应症,或者从处方药转换成了非处方药,则可以额外获得1年的数据保护期。新药在欧盟不需要提出专门的药品数据保护申请,在上市许可批准之后,自然获得。
欧盟试验数据保护制度并不按照药物类型区分市场独占期,分为以下几种[8]:
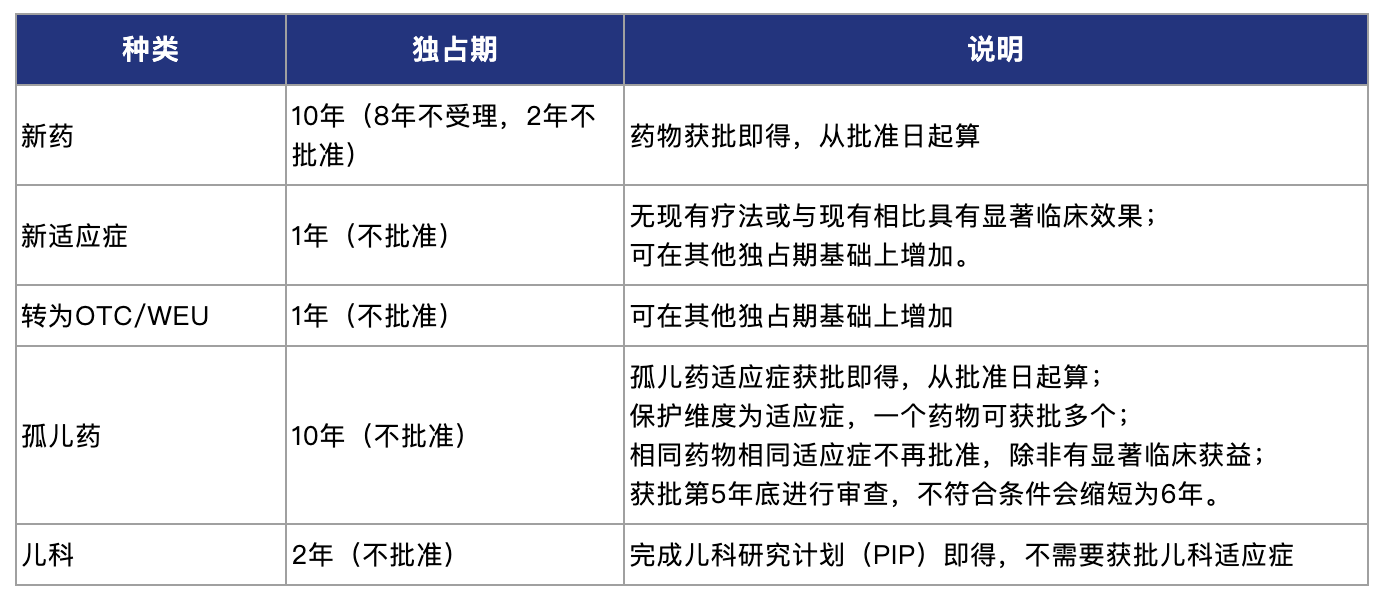
(2)药品市场排他期保护制度
1999年12月欧洲议会和理事会通过了《孤儿药条例》(EC 141/2000号条例),规定了获得孤儿药认定资格,对满足以下条件:1)必须用于治疗、预防或诊断危及生命或慢性衰弱的疾病;2)在欧盟的发病率不超过万分之五或药品上市销售收入不可能匹敌其研发所需的投入;3)没有令人满意的诊断、预防或治疗相关疾病的方法被批准,或如果这样的方法存在,但相关药物的显著效益受到相关疾病的影响。孤儿药上市许可获得批准之后,可获得10年的欧盟市场排他权,在此期间,直接竞争类似产品不能上市。如果在孤儿药认定审查期间,遵守儿科试验计划协议,市场排他权期限可再延长2年。
为进一步解释孤儿药定义及资格认定的标准,欧盟于2000年4月27日出台了EC 847/2000号条例,针对相似孤儿药的认定审批及临床优势问题作出详细解释。其中,相似孤儿药是指和已上市的孤儿药具有相似的活性物质,且用于相同的临床适应证的产品,主要从药物的分子结构特征、作用机制以及适应证三个角度。
三、对中国药企出海之启示
上述针对中国、美国与欧盟在药品排他保护制度的阐述,显示了国内外针对药品排他保护期的制度尚且存在一定的差异,欧美地区的药品排他保护制度体系相对更为复杂和完善,且各有侧重,详细了解相关国家和地区的药品排他保护制度对中国药企创新药与仿制药出海有着重要的指导意义。
对于中国创新药出海,国内药企应当在目标区域进行完善的专利布局,在此基础上,充分利用美国专利期限补偿(PTE)与调整(PTA)制度及欧盟的补充保证书制度(SPC),以便有效弥补药品上市审批所造成的药品专利保护期的损失,延长专利实际保护期。在专利审查过程中时密切关注专利审查进度,争取根据PTA制度获得因官方延误所导致的授权延误补偿。美欧的试验数据保护制度也不容忽视,中国药企应确保临床试验设计符合欧美标准,确保生成高质量的独占试验数据,并构建严格的数据管理体系,防止数据泄露,进而充分利用美欧地区的试验数据保护制度,阻却仿制药物的上市,获得更长的市场独占期。美欧针对孤儿药和儿科用药也均设立了完善的保障激励制度,除可以享受专利排他保护、试验数据保护制度外,国内药企可以通过聚焦细分适应症,以孤儿药切入低竞争赛道,争取获得相应的孤儿药、儿科用药的排他保护制度获得相应的排他保护期,从而获得更长的市场独占优势。
总之,中国药企出海需深入研究美欧药品排他保护制度,结合自身产品特性,合理运用欧美地区各项政策,制定针对性的出海策略,以便获得更长久的市场独占期,提升自身国际竞争力。
注释:
[1] 本文仅讨论发明专利。
[2]《专利法》第四十二条第三款:对在中国获得上市许可的新药相关发明专利,国务院专利行政部门应专利权人的请求给予专利权期限补偿。补偿期限不超过五年,新药批准上市后总有效专利权期限不超过十四年。
[3] https://www.fda.gov/drugs/development-approval-process-drugs/frequently-asked-questions-patents-and-exclusivity#informationlisted
[4] 梁志文:《药品数据保护的比较分析与立法选择》,载《政法论丛》2014年第5期,第80-88页。
[5] C.08.004.1. (1) Where a manufacturer files a new drug submission, an abbreviated new drug submission, a supplement to a new drug submission or a supplement to an abbreviated new drug submission for the purpose of establishing the safety and effectiveness of the new drug for which the submission or supplement is filed, and the Minister examines any information or material filed with the Minister, in a new drug submission, by the innovator of a drug that contains a chemical or biological substance not previously approved for sale in Canada as a drug, and the Minister, in support of the manufacturer's submission or supplement, relies on data contained in the information or material filed by the innovator, the Minister shall not issue a notice of compliance in respect of that submission or supplement earlier than five years after the date of issuance to the innovator of the notice of compliance or approval to market that drug, as the case may be, issued on the basis of the information or material filed by the innovator for that drug.
[6] Abbreviated New Drug Submission (ANDS) 是加拿大用于简化仿制药批准流程的监管程序。它允许仿制药制造商通过证明其产品与已批准的参照药物在生物等效性方面具有一致性,而无需进行新的临床试验来获得批准。
[7] 原文链接:EUR-Lex - 02009R0469-20190701 - EN - EUR-Lex (europa.eu)
[8] https://www.ema.europa.eu/en/documents/presentation/presentation-data-exclusivity-market-protection-orphan-and-paediatric-rewards-s-ribeiro_en.pdf

